
Restore Veto Property in Low Dose Aspirin/ASA Treated Preeclampsia Placenta Mesenchymal Stem Cells: Insights Into ASA-mediated Clinical Response*
Kimone A. Powell1, †, Lauren S. Sherman2, †, Bobak Shadpoor3, †, Shauna F. Williams1,* and Pranela Rameshwar2,*
1Department of Obstetrics, Gynecology and Reproductive Health, Rutgers New Jersey Medical School, Newark, NJ, USA
2Department of Medicine, Rutgers New Jersey Medical School, Newark, NJ, USA
3New Jersey Institute of Technology, Newark NJ, USA
E-mail: williash@njms.rutgers.edu; rameshwa@njms.rutgers.edu
*Corresponding Authors
†Equal contribution
Received 10 February 2024; Accepted 14 March 2024; Publication 20 April 2024
Preeclampsia (PE) is a pregnancy complication characterized by elevated blood pressure, proteinuria, and other laboratory abnormalities. PE affects 2–8% of pregnancies globally and can lead to preterm birth and other complications for the mother and fetus. Successful pregnancy depends on the ability of the mother’s immune system to tolerate the allogeneic fetus. However, in PE, this immune tolerance is exacerbated by inflammation. Transforming growth factor (TGF-) is important to retain an immune balance in healthy pregnancy. In PE, TGF- level is reduced, with an imbalance of the T-cell subset pool to favor inflammation. Omics studies by our group reported an increase in TGF- signaling when PE-derived placenta mesenchymal stem cells (P-MSCs) were treated with low dose aspirin (ASA). This correlated with increased cycling quiescence and epigenetic changes, resembling healthy P-MSCs. This study tested the hypothesis that ASA could restore the veto property of P-MSCs to mitigate inflammation. ASA (10 mM) treated P-MSCs from PE and healthy placentas increased TGF1 and its receptor. The ASA treated MSCs, when added as third-party cells to a one-way mixed lymphocyte reaction, suppressed T-cell proliferation. Prediction studies with omics data indicated that ASA-mediated TGF signaling could explain ASA-induced blunting of cell apoptosis. Together, the findings support ASA-mediated expression of TGF1 and its receptor on P-MSCs from PE to restore the ability to be licensed as immune suppressor to mitigate PE inflammation. The findings provide new insight into the benefit of ASA treatment for PE.
Preeclampsia (PE), presented as a multisystem pregnancy complication, continues to be clinical problem. PE is reported in 2–8 % of global pregnancies, and 5–8% in the United States. PE is the second most common cause of maternal morbidity and mortality worldwide [1–3]. In its most severe form, PE can cause fetal death, damage maternal end organs, and result in stroke or death [4].
In the United States, PE accounts for 6% of medically indicated preterm births [5, 6]. Maternal mortality from PE is generally higher in developing countries, although the number of cases continues to rise in the United States, reaching 18% in recent estimates [2, 6, 7]. The rate of PE increased by 25% between 1987 and 2004 [2]. Treating PE and related morbidity and mortality comes at great cost to the healthcare system. One study estimated that the cost of PE within 12 months of delivery was over 2 billion dollars in which most of the cost was attributable to preterm births [8].
PE is diagnosed with new onset elevated blood pressure after 20 weeks gestation or postpartum, along with other laboratory test abnormalities. The latter include proteinuria, thrombocytopenia, renal insufficiency, abnormal liver function, and/or pulmonary edema. Additional symptoms include new onset of headache and visual disturbances [8]. A prevailing view is that PE could be due to immune imbalance with increased proinflammatory CD4(+) T-cells and cytokines, with concomitant decrease of anti-inflammatory cells and mediators – regulatory T-cells (Tregs) and cytokines. Consequent to this imbalance are chronic inflammation, oxidative stress, predominance of proinflammatory cytokines, and autoantibodies [6].
The mechanisms leading to PE development are not entirely clear, but various theories have been proposed. The literature reported on shallow trophoblast invasion and inadequate spiral artery adaptation leading to hypoxia and oxidative damage [6]. Other proposed theories involve immune maladaptation and genetic imprinting, but more recent placental studies point to a key role for transforming growth factor (TGF-) over-expression during trophoblast invasion [9, 10]. As a result of placental dysfunction, inflammatory mediators are released into the maternal circulation, including free radicals, oxidized lipids, cytokines, and sFlt-1 (sVEGFR-1) [11]. These factors can cause generalized endothelial dysfunction, leading to the clinical symptoms of PE [12].
Although interventions such as medication, delivery, or magnesium sulfate can mitigate part of the associated complications and mortality of PE, the definitive treatment is limited to delivery [3]. Acetylsalicylic acid (aspirin, ASA) can reduce the risk of PE by 28% in women who are high risk and by 30% for women with average risk [13]. To date, the use of aspirin seems to be well tolerated with no report of risk to the mother and fetus [1, 4, 13]. The American College of Obstetricians and Gynecologists recommends ASA for women with more than one moderate risk factor or any high-risk factors starting between 12 to 28 weeks of gestation but optimally before 16 weeks [13].
Aspirin is a cyclooxygenase inhibitor with anti-inflammatory and antiplatelet properties, and used to prevent or delay the onset of PE [14]. Aspirin inhibits two cyclooxygenase isoenzymes (COX-1 and COX-2), which are necessary for prostaglandin biosynthesis [13]. In the past ASA has been used to prevent stillbirth, fetal growth restriction, preterm birth, and early pregnancy loss [13]. However, more recent practice used ASA to reduced PE incidence. However, there are insights into the benefits of ASA although its use in clinical practice varies [13].
Healthy pregnancy requires the maternal immune system to tolerate the fetus with a different major histocompatibility complex (MHC) [15–17]. This occurs when the activated maternal immune effectors are triggered by the trophoblast antigens to regulate the adaptive immune system [18]. Effectors in this immune balance include T-helper (Th), T-cytotoxic, T-Tregs, and B-cells [15, 16]. T-cell differentiation to specific subtypes is critical to the desired function to maintain immune balance during pregnancy. For instance, Th1 immunity is most crucial during peri-implantation to facilitate the invading trophoblasts [19]. After implantation, the dominant cells are Th2 to elicit anti-inflammatory response to protect the fetus and to facilitate fetal and placental development [20]. Th17 cells contribute to protecting against extracellular microbes during pregnancy [17]. In excess, Th17 immunity can lead to inflammation at the maternal-fetal interface [17]. Overall, dysregulation of Th cell immunity can result in complications to cause loss of pregnancy and preeclampsia [17].
PE subjects are presented with proinflammatory responses [6, 21]. Thus, it was assumed that treating PE subjects with ASA would mitigate the inflammatory response [4]. Studies have identified intracellular pathways associated with ASA treatment, such as increased TGF- signaling [6, 22, 23]. This particular response to ASA would benefit the inflammation in PE subjects since TGF- could increase regulatory T-cells to maintain fetal-maternal tolerance [24, 25]. The fetal-maternal surface has layers of placenta stem cells (P-MSCs) that share properties with mesenchymal stem cells (MSC) [14, 26]. The nomenclature used in this study to describe P-MSCs follows guidelines by the International Council for Commonality in Blood Banking Automation-International Society for Cell and Gene Therapy [27].
MSCs are multipotent cells found in various adult tissues such as bone marrow, adipose tissue and dental pulp [26]. MSCs can differentiate into cells of all germ layers [26]. MSCs, along with their secretome, can contribute to tissue regeneration [28–30]. MSCs can mediate both pro- and anti-inflammatory properties, depending on the milieu [31–33]. An inflammatory milieu could license MSCs to exert immune suppression with the relevant skewing of the immune response such as increased Tregs [34–36]. Despite several layers of P-MSCs at the surface of the placenta, it is unclear why the layers of P-MSCs cannot be licensed as immune suppressor to mitigate the enhanced inflammation in PE subjects [14].
We previously reported on ASA restoring the function of PE-derived P-MSCs (PE P-MSCs) by regulating cell cycle and resetting the epigenetic program of P-MSCs [37]. This study showed ASA restoring PE P-MSCs with the ability to respond to an inflammatory milieu for licensing into immune suppressor cells. The change in immune suppressor function was demonstrated with ASA-treated PE P-MSCs becoming veto cells when added as third-party cells to an allogeneic immune response [31]. We showed that ASA increased TGF-1 production and its receptor on P-MSCs from PE subjects, similar to healthy P-MSCs.
Dulbecco’s Modified Eagle Medium with High Glucose with Sodium Bicarbonate (DMEM), RPNI 1640, Triton X, Penicillin-Streptomycin, Fetal Bovine Sera (FBS) and RPMI-1640, sodium heparin, dexamethasone, -glycerophosphate, silver nitrate, Ficoll Hypaque, and paraformaldehyde were purchased from Sigma Aldrich (St Louis, MO), FBS were heat inactivated for 45 min at 56C. L-Glutamine, 200 mM, 1x phosphate buffered saline (PBS), and Trypsin/EDTA from Life Technologies (Carlsbad, CA); 4,6-diamidino-2-phenyl indole, dilactate (DAPI) from Molecular Probes-Life Technologies.
The following antibodies were purchased from BD Pharmingen: APC – mouse anti-human CD105, PerCP-Cy5.5 – anti-human CD90, FITC – anti-human CD73, PE – anti-human CD105, FITC – anti-mouse IgG, PE – anti-mouse IgG1, PerCP-Cy5.5 – anti-mouse IgG1. PE-goat anti-rabbit was purchased from Abcam (Waltham, MA); anti-TGFR1 from Invitrogen (Carlsbad, CA) and TGF1 from R&D Systems (Minneapolis, MN).
CCL-64 mink lung epithelial cell was purchased from American Type Culture Collection. CCL-64 culture was previously described [38]. Briefly, the cells were cultured in RPMI 1640 with 10% FBS, Hepes (0.015M), 1% Penicillin-Streptomycin (stock = 5,000 U Penicillin and 5,000 g Streptomycin), and 2 mM L-Glutamine.
This study was approved by the Rutgers Institutional Review Board (Rutgers University, NJ). All participants signed the informed consent.
The study included subjects with singleton term gestations from normal physiological control pregnancies and those complicated by PE. Individuals were recruited to the PE group if they met the traditional diagnostic criteria for PE, including hypertension (greater than 140 mm Hg systolic or 90 mm Hg diastolic blood pressure) on two instances at least 4 hours apart, proteinuria, or indicators of severe disease outlined by the American College of Obstetricians and Gynecologists [8]. Women and fetuses with unrelated comorbidities (e.g. diabetes, autoimmune disease; fetal congenital or chromosomal abnormalities) were excluded.
Subjects with healthy physiologic pregnancies were included in the control group. Individuals were excluded from recruitment based on the following criteria: women with serious co-morbidities, such as pre-existing hypertension, renal disease, diabetes, thyroid disorders, autoimmune or collagen vascular disease, fetal congenital or chromosomal anomalies, placental abnormalities such as placenta previa, intrauterine infection such as chorioamnionitis, and other notable infections such as HIV, hepatitis B/C, or COVID.
P-MSCs were expanded from human placentas as described [14, 37]. Briefly, placentas were obtained from term vaginal or cesarean delivery and then immediately placed in a sterile container for transport to the laboratory for processing under a laminar flow hood. The placentas were washed several times with PBS containing P-S. We stopped washing when there was visually negligible blood in the tissue. The placentas were manually dissected by peeling apart the amniotic and chorionic membranes, after which the chorionic membrane (CM) was minced into 2–5 mm pieces using sterile forceps and surgical scissors. The minced pieces of tissue were placed into 100 mm tissue culture-treated dishes (Falcon) containing P-MSC media (DMEM with 10% FBS, P-S, and L-glutamine). The dishes were incubated at 37C with 5% CO. After 3 days, 50% of the media was removed and replaced with fresh P-MSC media. After seven days, the minced tissues were removed. At this time, there were adherent cells that were spindle shaped. At weekly intervals thereafter, approximately 50% of the culture media were replaced until 70–80% confluence for continued passaging. The passaged cells contained media – 50% fresh and 50% from previous culture plates, as described [39]. After three passages P-MSCs were characterized by multilineage differentiation and flow cytometry for established P-MSC markers (Figure 1).
Figure 1 Characterization of P-MSCs. A) Representative morphology of passaged P-MSCs. B) P-MSCs from healthy and PE placentas were analyzed by flow cytometry with anti- CD90, CD73 and CD105. Top panels represent isotype control. Representative scatter images are shown for ASA-treated P-MSCs (middle panels) and vehicle (lower panels). C) P-MSCs were induced with adipogenic and osteogenic media. Shown are representative images for each induction.
At 70% confluence, P-MSCs were de-adhered with Trypsin-EDTA solution and then placed into DMEM with 10% FCS. The cells suspension was incubated for 30 min at room temperature to allow for re-expression of surface proteins that might have been destroyed by trypsin. The cells were centrifuged and then resuspended in sera-free DMEM followed by incubation with primary conjugated antibodies (CD73-FITC, CD90-PerCP-Cy5.5, CD105-PE, CD34-FITC, CD45-PE) at 1/20 final dilution. Cells were read immediately and analyzed by flow cytometry (FACS Caliber; Becton Dickinson, Franklin Lakes, NJ).
Freshly prepared ASA, diluted in media, was used in tissue culture assays. P-MSCs (80% confluence) from placentas of PE or healthy pregnancies were cultured for 24 h or 48 h in MSC media containing 1 mM or 10 mM aspirin. These concentrations of ASA are consistent with literature to recapitulate the physiologic range of low dose ASA while avoiding cell-death due to aspirin’s acidity [14, 37, 40–43]. Furthermore, using these guidelines, our group previously reported on this concentration in studies relevant to the present study [14, 37].
P-MSCs were seeded in 6-well plates at 3 10 cells/well in MSC media containing 10 mM ASA or vehicle. The cells were incubated at 37C. At 24 and 48 h, the media were aspirated followed by washing of the cells washed with PBS. The cells were fixed with 3.7% PFA for 20 min at room temperature, washed with PBS, and then permeabilized with 0.2% Triton-X 100 for 5 min at room temperature. The permeabilized cells were incubated with blocking solution (0.5% Triton X-100, 0.5% BSA) for 1 h while shaking at room temperature. After this, the cells were incubated overnight at 4C with anti-TGFR1 at 1:1000 in blocking solution. The cells were washed three times with PBS with each wash occurring for five mins. The primary antibody was detected with Alexa Fluor anti-rabbit IgG at 1:500. After 2 h at room temperature, the cells were counterstained by incubating with DAPI at 1:1000 for 10 mins. The cells were washed with PBS and imaged immediately with the EVOS FL Auto 2 microscope (Fisher Scientific).
Active TGF 1 quantification was based on growth inhibition of CCL-64 cells, as described [38]. The CCL-64 cells were plated in 24-well tissue culture plates at 10 cells/mL in 500 L of RPMI 1640 with 10% FCS. The plates were incubated at 37C and 5% CO. The next day, standard TGF 1 was added to the cells at concentrations ranging between 100 pM and 0.01pM. Test samples (500 L) were added in triplicates to wells. The plates were incubated at 37C and 5% CO for 72 h. The total number of viable cells were assessed by trypan blue exclusion with counting on a hemacytometer. Samples with TGF- concentrations 20 ng/ml were retested with neutralizing anti-hTGF- to confirm specificity of the cytokine.
Whole blood was obtained from two donors into heparinized tubes. The peripheral blood mononuclear cells (PBMCs) were collected via Ficoll Hypaque (Sigma Aldrich) density gradient centrifugation following the manufacturer’s protocol. One donor’s PBMCs were designated as the stimulator cells: these cells were resuspended at 10 10 in RPMI and irradiated with 5000 rads from a Cesium (Cs) source prior to resuspending at 2 10/ml. PBMCs from the second donor were designated as the responder cells and resuspended at 2 10. PBMCs from the two donors were plated in equal numbers with or without addition of P-MSCs and incubated at 37C and 5% CO for 72 hours. At 72 h, the cells were pulsed with 1 Ci of tritiated thymidine. After 16 h, the cells were harvested on a Cambridge Technology PHD Cell Harvester. The filters were assessed for tritiated thymidine incorporation on a Beckman Coulter LS6500 Liquid Scintillation Counter.
Adipogenic differentiation used the Differentiation Bullet Kit from Cambrex BioScience (Walkersville, MD), following manufacturer’s instructions, and previously described [31]. Briefly, MSCs (5 10), were added to 6-well tissue culture plates. At confluence, adipogenic differentiation was performed with 3 cycles of induction/maintenance media. Each cycle consisted of 3-day culture in Adipogenic Induction Medium followed by 1–3 days with Adipogenic Maintenance Medium. Control wells were similarly cultured except with Adipogeinc Maintenance Medium. After the 3 cycles, cells were grown in Adipogenic Maintenance Media for 7 days, with the media replaced every 3 days. After induction, cells were washed with PBS, fixed with 10% formalin, and stained with Oil Red O and counterstained with hematoxylin.
Osteogenic differentiation was plated as for adipogenic differentiation. The cells were induced in DMEM with 10% FCS, 100 nM Dexamethasome, 10 mM -glycerophosphate, and 50 M L-ascorbic acid-2-phosphate. Differentiation media was replaced every 3–4 days for 3 weeks. Controls wells were similarly cultured in MSC media. Osteogenic differentiation was assessed by mineral deposits with von Kossa stain. This was done by first fixing with 10% formalin, stained for 10 min in the dark with 2% silver nitrate solution, washed and exposed for 15 min to light.
Experimental points were compared using Mann Whitney U test and Student’s t-test.
P-MSCs were cultured from the CM of healthy pregnancies or from subjects with preeclampsia, shown in Table 1, and with P-MSCs from previous studies [14, 37]. To eliminate residual hematopoietic cells and fibroblasts from cultured P-MSCs, we characterized the cells after passage 3. At this time, P-MSCs from healthy and PE subjects were uniformly plastic-adherent and characteristically spindle-shaped (Figure 1A). Phenotypic studies by flow cytometry indicated that the cells before and after ASA treatment were positive for MSC markers – CD73, CD90, and CD105, and negative for CD34, and CD45 (Figure 1B). The cells were also negative for CD34 and CD45, which eliminated contamination by residual hematopoietic cells. The MSCs from both healthy and PE subjects showed multilineage differentiation to osteogenic and adipogenic cells (Figure 1C).
Table 1 Maternal demographics and gestational age
| Healthy (N 7) | Preeclamptic (N 6) | |
| Maternal age (yrs) | 32 (21–42) | 25.2 (16–34) |
| Gestational age (wks) | 39 (39–40.5) | 39 (35.4–40.4) |
| Shown are the total number of placentas acquired for the study. Also shown is the gestational age indicating term delivery. | ||
RNA-Seq analyses supported increased TGF1 signaling in ASA-treated PE-derived P-MSCs [14]. MSCs can mitigate inflammation by licensing as immune suppressor cells [31]. We therefore asked if ASA can restore the ability of P-MSCs from PE to become immune suppressor cells by enhanced response to TGF-1 [44]. First, we asked if ASA increase TGF-1 receptor expression on P-MSCs. We cultured P-MSCs from PE placentas with 10 mM ASA. After 24 h, we evaluated the cells for TGF receptor by immunocytochemistry (Figure 2). The results showed increased labeling with ASA treatment, as compared to vehicle, indicating increased TGF receptor expression.
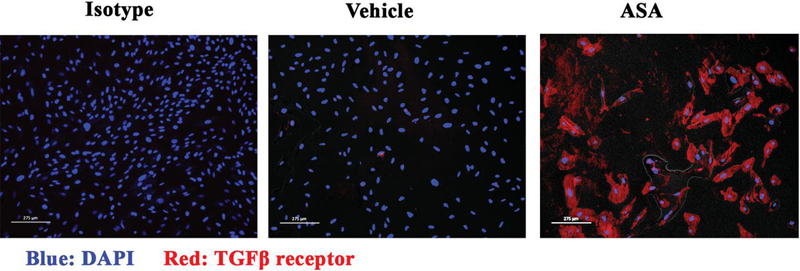
Figure 2 ASA-induced TGF receptor on P-MSCs. P-MSCs from chorionic membrane were treated with 10 mM ASA or vehicle for 24 h. The cells were labeled with anti-TGF- receptor (red) and then counter stained with DAPI (blue) for nuclear identification. The images represent 3 biological replicates.
ASA induced the levels of membrane TGF1 receptor (Figure 2). We therefore asked if the P-MSCs could be stimulated with TGF-1 through autocrine mechanisms. This could be achieved if P-MSCs produce TGF-1. To this end, we stimulated P-MSCs from healthy and PE with 1 mM ASA or vehicle. After 24 h, cell-free media were analyzed for active TGF1 in a bioassay with CCL-64. We established a standard curve with known concentrations of TGF-1 (Supplemental Figure 1). To ensure that residual ASA in the media did not affect the viability of CCL64, we conducted the assay with media from vehicle supplemented with ASA. The addition of ASA to vehicle media did not alter CCL 64 viability (Supplemental Figure 2). As compared to baseline TGF-1, ASA induced significant (p 0.05) increase in the production of active TGF-1 (Figure 3A). TGF-1 in the media was confirmed by performing the assay in parallel with neutralizing anti-TGF-1 (Figure 3A, right bar). In summary, ASA induced the production of TGF-1 in P-MSCs.

Figure 3 ASA induced production of TGF-1 in P-MSCs. A) P-MSCs were treated with vehicle or 1 mM ASA. After 24 h, the media were collected and then quantitated for active TGF- in a bioassay with CCL64 cells. Parallel quantitation was performed in the presence of neutralizing anti-TGF-1. The data represent the mean level SD, n 4. B) RNA-Seq data from previous studies was analyzed by IPA, ASA vs. vehicle [14]. *p 0.05 vs. media or neutralizing antibody.
We used the published data in GEO to determine if the changes in gene expression could predict TGF-1 signaling using Ingenuity Pathway Analyses, (IPA; Qiagen) [14]. IPA analyses of ASA vs. vehicle treatment indicated an increase in TGF- signaling with complexing of Smad4 and Smad 2/3 (Figure 3B). Interestingly, the pathway also showed TGF- signaling inhibiting apoptosis. This correlated with Smad 4 showing increased anti-apoptotic Bcl2 (Figure 3B). Overall, the predicted in silico analyses of pervious RNA-Seq data, which showed consistency with the increased levels of TGF-1 production and its receptor (Figures 2 and 3A) [14].
MSCs can be licensed to exert immune suppressor function as third party cells in one-way and two-way MLR [31, 32]. We therefore asked if the ASA-mediated transition of PE P-MSCs to cycling quiescent cells and epigenetic changes could also allow the PE P-MSCs to be licensed as immune suppressor cells [14, 37]. If so, this could explain why ASA showed clinical benefit for PE.
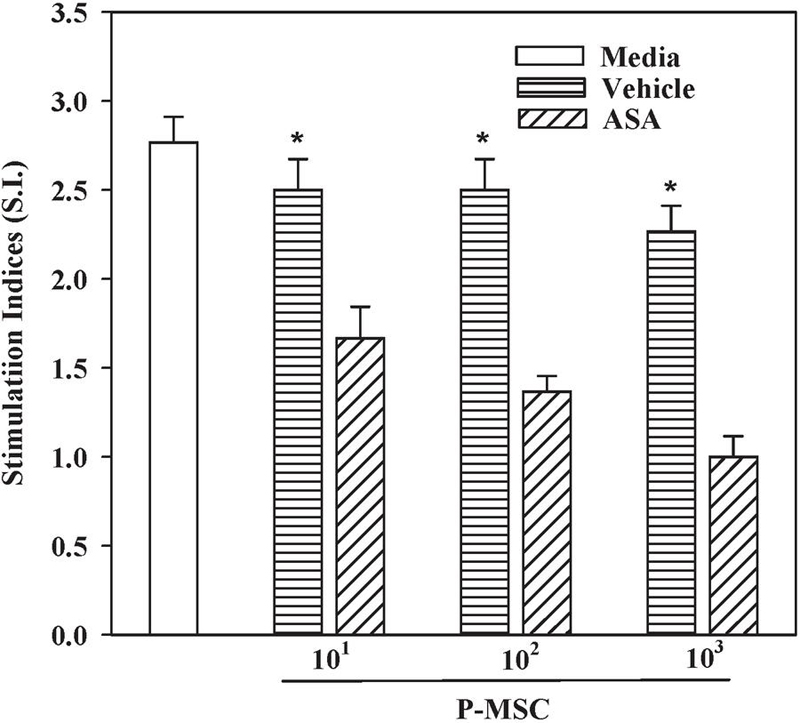
Figure 4 ASA-mediated licensing of P-MSCs. One-way MLR was established with peripheral blood from two allogeneic donors and third party P-MSCs from PE subjects. The P-MSCs were stimulated with vehicle or 1 mM ASA for 24 h. The data are presented as mean stimulation indices (S.I.SD, n 3). * p 0.05 vs. ASA.
We established a one-way MLR with P-MSCs from PE that were treated with vehicle or 1 mM ASA. The stimulation indices in the assay indicated significant (p 0.05) suppression when the PE P-MSCs were treated with ASA, as compared to vehicle treated P-MSCs (Figure 4). The degree of proliferative suppression was proportional to the number of MSCs. In summary, ASA enhanced the capacity of P-MSCs from PE placenta to inhibit allogeneic responses of peripheral blood mononuclear cells.
PE is generally considered an inflammatory disorder of pregnancy with ASA provided to mitigate the response via its pan-anti-inflammatory property, hence its widespread use in pregnancy. One of the indications for ASA includes its use for recurrent pregnancy loss, systemic lupus erythematosus, and preeclampsia [13]. However the mechanisms by which ASA elicits positive clinical response is a subject of experimental investigations. Previous work from our group has provided insights into the mechanisms by which ASA could exert its clinical response in PE [14, 37]. These include reduced proliferation, enhanced angiogenesis, and changes in the epigenetic landscape. There was also evidence of reduced inflammation based on analyses of the omics studies [14]. Together, these studies led us to determine if the effects of ASA treatment could act indirectly through P-MSCs; specifically by restoring the ability of PE-derived P-MSCs to mitigate inflammation. We report on a systematic set of studies showing ASA restoring PE-derived P-MSCs to become veto cells, which exert immune suppression [31].
First, we showed ASA increasing TGF- receptor and TGF-1 production (Figures 2 and 3). These findings were consistent with IPA analyses of previous RNA-Seq data, showing an increase in TGF-1 signaling (Figure 3B). Previous studies predicted an increase in cell senescence in vehicle treated P-MSCs from PE placenta [37]. We now showed ASA with the potential to increase the anti-apoptotic Bcl2. These findings indicated that P-MSCs could be restored to mitigate the inflammation linked to PE.
Using allogeneic the proliferative studies, we noted that ASA treated P-MSCs from PE placentas could suppress the inflammation (Figure 4). This was an interesting finding due to insights into how ASA could be able to mitigate inflammation in PE. The MLR response indicated that ASA provided P-MSCs from PE subjects with the ability to be licensed as immune suppressor cells, as reported for veto activity [31]. TGF-1 can induce regulatory T-cells and this could occur from the release of this cytokine from MSC [44]. By extrapolation, it appears that ASA mitigation of PE could in part occur by induced TGF-1, which can change the immune landscape. Indeed, ASA has been reported to change the immune cell balance in PE subjects [45, 46]. Although not investigated, future studies could study the immune cell distribution in the MLR shown in Figure 4. Other studies include if TGF-1 is limited to autocrine activation on P-MSCs or if this cytokine could influence T-cell differentiation to skew the reaction from pro-inflammatory to immune suppressor cells.
As discussed in the previous paragraph, restoring P-MSCs from PE to mitigate inflammation should result in their ability to increase regulatory T-cells [44]. Furthermore, we previously reported on ASA mediating the reorganization of the epigenetic program of PE P-MSCs through the base excision repair (BER) and nucleotide excision repair (NER) pathways [37]. This study tested the hypothesis that ASA enhanced TGF- signaling pathway. Together, the findings, combined with the two previous cited reports on RNA-Seq data, suggested that TGF-1 signaling could be increased with ASA treatment [14].
Prior studies of TGF- in PE have shown mixed results [24, 47, 48]. In most cases, there are differences in the tissues used or the demographics of the subjects. Many of these studies measured TGF- including both the latent and active forms of the cytokine. The present report clarifies the response of ASA since we assessed the active cytokine from P-MSCs. These findings are consistent with ability of ASA treated P-MSCs to act as veto cells. Indeed, the MLR studies using ASA-treated P-MSCs from PE subject were important to test if ASA can restore normal physiological pregnancy, which requires tolerance between the maternal and fetal immune systems. The maternal immune system undergoes many changes to adapt to the growing fetus which may express paternal antigens. TGF plays a role in this process because it is involved in regulating immune cell function. In preeclampsia, TGF- expression was found to be significantly reduced in one study [24].
Further studies are necessary to gain a complete understanding of how ASA mitigates PE pathology. Specifically, a deeper investigation into the downstream effects of altered TGF- signaling in PE-MSCs is imperative. Additionally, it is essential to observe the role of SMAD4, a downstream mediator of TGF- signal transduction, to determine whether rescuing TGF- signaling is necessary and/or sufficient to restore PE-MSCs towards healthy function. Overall, this study brings us closer to discerning how ASA functions to mitigate PE.
Overall, the data support our hypothesis that the ASA-treated PE P-MSCs can be licensed as third-party immune suppressing cells. These cells were able to function as normal MSCs and effectively suppress cellular proliferation in an inflammatory environment. These findings suggest that ASA treatment can potentially enhance the therapeutic benefits of MSCs in the treatment of inflammation. These findings have potential utility in other obstetric or non-obstetric diseases which are caused by inflammation.
The studies were supported by a discretionary fund to PR.
KAP collected the placentas, expanded P-MSCs, performed the experiment and wrote the first draft of the manuscript; LSS cultured the P-MSCs, wrote the manuscript, analyzed the data and performed the experiments; BS cultured the P-MSCs, performed the experiments; analyzed the data and edited the manuscript; SW contributed to the concepts, edited the manuscript and analyzed the data; PR contributed to the concepts, edited and approved the final version of the manuscript, and analyzed the data.
*This work was conducted at Rutgers New Jersey Medical School, Departments of Medicine and Obstetrics, Gynecology and Reproductive Health
[1] Force USPST, Davidson KW, Barry MJ, Mangione CM, Cabana M, Caughey AB, et al. Aspirin Use to Prevent Preeclampsia and Related Morbidity and Mortality: US Preventive Services Task Force Recommendation Statement. J Am Med Assoc. 2021;326:1186–1191.
[2] Kuklina EV, Ayala C, Callaghan WM. Hypertensive disorders and severe obstetric morbidity in the United States. Obstet Gynecol. 2009;113:1299–306.
[3] Brooks SA, Martin E, Smeester L, Grace MR, Boggess K, Fry RC. miRNAs as common regulators of the transforming growth factor (TGF)-beta pathway in the preeclamptic placenta and cadmium-treated trophoblasts: Links between the environment, the epigenome and preeclampsia. Food Chem Toxicol. 2016;98(Pt A):50–7.
[4] Wang Y, Guo X, Obore N, Ding H, Wu C, Yu H. Aspirin for the prevention of preeclampsia: A systematic review and meta-analysis of randomized controlled studies. Front Cardiovasc Med. 2022;9:936560.
[5] Davidson KW, Barry MJ, Mangione CM, Cabana M, Caughey AB, Davis EM, et al. Aspirin use to prevent preeclampsia and related morbidity and mortality: US Preventive Services Task Force recommendation statement. J Am Med Assoc. 2021;326:1186–91.
[6] Cornelius DC. Preeclampsia: From Inflammation to Immunoregulation. Clin Med Insights Blood Disord. 2018;11:1179545X17752325.
[7] Tsakiridis I, Giouleka S, Arvanitaki A, Giannakoulas G, Papazisis G, Mamopoulos A, et al. Gestational Hypertension and Preeclampsia: An Overview of National and International Guidelines. Obstet Gynecol Surv. 2021;76:613–633.
[8] Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin Summary, Number 222. Obstet Gynecol. 2020;135:1492–1495.
[9] Simpson H, Robson SC, Bulmer JN, Barber A, Lyall F. Transforming growth factor beta expression in human placenta and placental bed during early pregnancy. Placenta. 2002;23:44–58.
[10] Dekker GA, Sibai BM. Etiology and pathogenesis of preeclampsia: current concepts. Am J Obstet Gynecol. 1998;179:1359–1375.
[11] Jena MK, Sharma NR, Petitt M, Maulik D, Nayak NR. Pathogenesis of preeclampsia and therapeutic approaches targeting the placenta. Biomolecules. 2020;10:953.
[12] Atallah A, Lecarpentier E, Goffinet F, Doret-Dion M, Gaucherand P, Tsatsaris V. Aspirin for Prevention of Preeclampsia. Drugs. 2017;77:1819–1831.
[13] ACOG Committee Opinion No. 743: Low-Dose Aspirin Use During Pregnancy. Obstet Gynecol. 2018;132:e44–e52.
[14] Romagano MP, Sherman LS, Shadpoor B, El-Far M, Souayah S, Pamarthi SH, et al. Aspirin-Mediated Reset of Preeclamptic Placental Stem Cell Transcriptome – Implication for Stabilized Placental Function. Stem Cell Rev Rep. 2022;18:3066–3082.
[15] Abu-Raya B, Michalski C, Sadarangani M, Lavoie PM. Maternal Immunological Adaptation During Normal Pregnancy. Front Immunol. 2020;11:575197.
[16] Zhang YJ, Shen L, Zhang T, Muyayalo KP, Luo J, Mor G, et al. Immunologic Memory in Pregnancy: Focusing on Memory Regulatory T Cells. Int J Biol Sci. 2022;18:2406–2418.
[17] Wang W, Sung N, Gilman-Sachs A, Kwak-Kim J. T Helper (Th) Cell Profiles in Pregnancy and Recurrent Pregnancy Losses: Th1/Th2/Th9/Th17/Th22/Tfh Cells. Front Immunol. 2020;11:2025.
[18] Warning JC, McCracken SA, Morris JM. A balancing act: mechanisms by which the fetus avoids rejection by the maternal immune system. Reproduction. 2011;141:715–724.
[19] Robertson SA, Moldenhauer LM. Immunological determinants of implantation success. International J Dev Biol. 2014;58:205–217.
[20] Kaicker A. Immune cells at the maternal-fetal interphase: Role in implantation and establishment of tolerance. Journal of Applied Biology and Biotechnology. 2023;11:45–50.
[21] Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW, Jr., Wallace K, et al. The role of inflammation in the pathology of preeclampsia. Clin Sci (Lond). 2016;130:409–19.
[22] Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. TGF-beta – an excellent servant but a bad master. J Transl Med. 2012;10:183.
[23] Zhang Y, Alexander PB, Wang XF. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb Perspect Biol. 2017;9:a022145.
[24] Yang D, Dai F, Yuan M, Zheng Y, Liu S, Deng Z, et al. Role of Transforming Growth Factor-beta1 in Regulating Fetal-Maternal Immune Tolerance in Normal and Pathological Pregnancy. Front Immunol. 2021;12:689181.
[25] Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol. 2017;9:a022236.
[26] Sherman LS, Shaker M, Mariotti V, Rameshwar P. Mesenchymal stromal/stem cells in drug therapy: New perspective. Cytotherapy. 2017;19:19–27.
[27] Viswanathan S, Ciccocioppo R, Galipeau J, Krampera M, Le Blanc K, Martin I, et al. Consensus International Council for Commonality in Blood Banking Automation-International Society for Cell & Gene Therapy statement on standard nomenclature abbreviations for the tissue of origin of mesenchymal stromal cells. Cytotherapy. 2021;23:1060–1063.
[28] Barry FP, Murphy JM. Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004;36: 568–584.
[29] Helmy KY, Patel SA, Silverio K, Pliner L, Rameshwar P. Stem cells and regenerative medicine: accomplishments to date and future promise. Ther Deliv. 2010;1:693–705.
[30] Eljarrah A, Gergues M, Pobiarzyn PW, Sandiford OA, Rameshwar P. Therapeutic Potential of Mesenchymal Stem Cells in Immune-Mediated Diseases. Adv Exp Med Biol. 2019;1201:93–108.
[31] Potian JA, Aviv H, Ponzio NM, Harrison JS, Rameshwar P. Veto-like activity of mesenchymal stem cells: functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol. 2003;171:3426–3434.
[32] Kang HS, Habib M, Chan J, Abavana C, Potian JA, Ponzio NM, et al. A paradoxical role for IFN-gamma in the immune properties of mesenchymal stem cells during viral challenge. Exp Hematol. 2005;33: 796–803.
[33] Jiang W, Xu J. Immune modulation by mesenchymal stem cells. Cell Prolif. 2020;53:e12712.
[34] Wang M, Yuan Q, Xie L. Mesenchymal Stem Cell-Based Immunomodulation: Properties and Clinical Application. Stem Cells Int. 2018;2018: 3057624.
[35] Salari V, Mengoni F, Del Gallo F, Bertini G, Fabene PF. The Anti-Inflammatory Properties of Mesenchymal Stem Cells in Epilepsy: Possible Treatments and Future Perspectives. Intl J Mol Sci. 2020;21:9683.
[36] Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736.
[37] Krishnamoorthy K, Sherman LS, Romagano MP, El Far M, Etchegaray JP, Williams SF, et al. Low dose acetyl salicylic acid (LDA) mediates epigenetic changes in preeclampsia placental mesenchymal stem cells similar to cells from healthy pregnancy. Placenta. 2023;137:49–58.
[38] Rameshwar P, Narayanan R, Qian J, Denny TN, Colon C, Gascon P. NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: an inflammatory response beyond the realm of homeostasis. J Immunol. 2000;165:2271–2277.
[39] Sandiford OA, Donnelly RJ, El-Far MH, Burgmeyer LM, Sinha G, Pamarthi SH, et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021;81:1567–1582.
[40] Hao W, Shi S, Zhou S, Wang X, Nie S. Aspirin inhibits growth and enhances cardiomyocyte differentiation of bone marrow mesenchymal stem cells. Eur J Pharmacol. 2018;827:198–207.
[41] Wang Y, Chen X, Zhu W, Zhang H, Hu S, Cong X. Growth inhibition of mesenchymal stem cells by aspirin: involvement of the WNT/beta-catenin signal pathway. Clin Exp Pharmacol Physiol. 2006;33:696–701.
[42] Fan W, Li J, Chen J, Zhu L, Wang Y, Sun B, et al. Aspirin inhibits the proliferation of synovium-derived mesenchymal stem cells by arresting the cell cycle in the G0/G1 phase. Am J Transl Res. 2017;9:5056–5062.
[43] Cao Y, Xiong J, Mei S, Wang F, Zhao Z, Wang S, et al. Aspirin promotes bone marrow mesenchymal stem cell-based calvarial bone regeneration in mini swine. Stem Cell Res Ther. 2015;6:210.
[44] Patel SA, Meyer JR, Greco SJ, Corcoran KE, Bryan M, Rameshwar P. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: role of mesenchymal stem cell-derived TGF-beta. J Immunol. 2010;184:5885–5894.
[45] Rambaldi MP, Weiner E, Mecacci F, Bar J, Petraglia F. Immunomodulation and preeclampsia. Best practice Res Clin Obstetrics Gynaecol. 2019;60:87–96.
[46] Ai-ris YC, Smith LA, Karumanchi SA. Review of the immune mechanisms of preeclampsia and the potential of immune modulating therapy. Human Immunol. 2021;82:362–370.
[47] Molvarec A, Szarka A, Walentin S, Beko G, Karadi I, Prohaszka Z, et al. Serum leptin levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in normal pregnancy and preeclampsia. Reprod Biol Endocrinol. 2011;9:124.
[48] Yu L, Kuang LY, He F, Du LL, Li QL, Sun W, et al. The Role and Molecular Mechanism of Long Nocoding RNA-MEG3 in the Pathogenesis of Preeclampsia. Reprod Sci. 2018;25:1619–1628.
International Journal of Translational Science, Vol. 1, 133–152.
doi: 10.13052/ijts2246-8765.2024.025
© 2024 River Publishers