A Platform to Establish a Working Hierarchy of Glioblastoma Multiforme Cells: Implication on Cancer Cell-microenvironment Interaction and Response to Drugs
Vibha Harindra Savanur1, 2, Anushka Sarkar1, 2, Andrew Petryna1, 2, Ky Nguyen1, 2, Jesus Benites-Sandoval1, Marina Gergues1, 2, Arash Hatefi3 and Pranela Rameshwar1,*
1Rutgers New Jersey Medical School, Newark, NJ, USA
2Rutgers School of Graduate Studies at New Jersey Medical School, Newark, NJ, USA
3Department of Pharmaceutics, Rutgers University, Piscataway, NJ 08854, USA
E-mail: rameshwa@njms.rutgers.edu
*Corresponding Author
Received 28 April 2024; Accepted 28 June 2024
Abstract
Glioblastoma multiform (GBM), a grade IV glioma, is the most common and aggressive cancer in the central nervous system. Current treatment for GBM includes surgical resection, radiation, and the frontline DNA alkylating drug, temozolomide (TMZ). The current median survival for GBM patients is about 14.5 months with 5% patients surviving up to 5 years. We propose that discerning distinct subsets within heterogeneous GBM will provide avenues for research to improve new therapies. We used different methods to isolate GBM cell subsets. These include stable transfectants of GBM cell lines with a lentiviral system in which green fluorescence protein (GFP) is regulated with tandem repeats of Oct4a and Sox2 response elements. Parallel studies with a plasmid using the full-length regulatory region of Oct4a indicated reduced efficiency in separating cell subsets, relative to SORE6-GFP lentivirus. Stem cell-linked gene expressions and function studies such as ALDH1, tumorsphere and in vivo passaging of GFP hi subsets confirmed the presence of cancer stem cells (CSCs). We also studied a more efficient method that could be relevant for primary GBM cells. We selected tumorspheres by plating heterogeneous GBM cells and then serially passaged the spheres. Studies for stem cell genes indicated that this method could be used for primary GBM cells. Overall, this study provided insights into methods to isolate GBM subsets, including primary GBM cells. The advantages of the methods are discussed.
Keywords: Cancer stem cells, brain, glioblastoma.
Introduction
Glioblastoma Multiform (GBM), a grade 4 astrocytoma, continues to be the most aggressive and common form of primary malignant glioma in humans [1–3]. Primary (de novo) GBM showed no evidence of pre-existing lower grade glioma [4]. Secondary GBM develops from the progression of a lower grade astrocytoma into metastatic tumor. Although there is no definite cause for cancer driver mutations in GBM, ionizing radiation, environmental factors, random gene mutations or germline pre-dispositions could be involved in tumor development [5]. Primary and secondary GBM differ with respect to molecular pathways, rate of tumor progression, treatment strategies, and clinical outcomes.
The World Health Organization classified gliomas into four malignant grades, based on histology. Grades I and II are non-malignant while Grades III and IV are malignant. GBM mostly occurs in individuals, age 45–70 years, although more recent information reported on GBM at 35 years [6]. The area of the brain affected by GBM could result in discernible symptoms such as numbness, loss of vison, weakness, and seizures [7]. The sensitivity of Magnetic Resonance Imaging (MRI) can detect GBM by identifying enhanced lesions with necrosis at the center. However, due to GBM heterogeneity, the disease can be presented differently, which makes diagnosis difficult until the cancer has advanced [8].
Standard care for GBM includes surgical resection of the tumor, followed by radiation and chemotherapy with the DNA alkylating agent, temozolomide (TMZ) [9]. Despite treatment, the prognosis for GBM is poor with less than 14 months survival, and less than 5% surviving up to 5 years [10]. Therefore, it is critical to identify new treatments for GBM, perhaps in combination with TMZ. Success can be improved by research aimed to isolate GBM cell subsets.
GBM cells can acquire mutations to resist TMZ [11, 12]. MGMT is the most important contributor of TMZ resistance due to its ability to repair DNA mismatch [13, 14]. MGMT removes the methyl group in O6-methylguanine created by TMZ to prevent DNA damage, resulting in poor drug response. Another impediment is the blood brain barrier (BBB), which prevents most drugs from crossing into the brain.
Sonic hedgehog (SHH) pathway has been implicated in GBM biology [15–17]. MicroRNA9 (miR9) suppresses PTCH1 translation resulting in Gli 1 activation [14]. Recurrence of GBM has been partly linked to resistant cancer stem cells (CSCs) [18, 19]. CSCs are mostly in cellular quiescence where they survive as dormant cancer cells with the ability to initiate metastatic cancer [20, 21]. CD133 and aldehyde dehydrogenase (ALDH) activity have been reported to identify CSCs with GBM [22]. However, other reports provide doubts on the specificity of these two markers for GBM CSCs [23–25]. CD133 is not specific for cancer cells since it is also expressed on healthy stem cells, and other cells [24, 25]. Similarly, ALDH is active in CSCs and cancer progenitors, healthy stem cells, and other malignant cells [26, 27].
CSCs from breast cancer were selected with a plasmid containing the full 5 regulatory region of the stem cell gene, Oct4a linked to green fluorescence protein (GFP) [28]. In this study, we applied a similar principle with a lentivirus containing tandem repeats of Sox2 and Oct4a linked to GFP (SORE6-GFP) [29–31]. Regardless of the method, developing a hierarchy of GBM would have great impact to advance the research on GBM since it will be possible to evaluate how the cells react to microenvironmental factors.
Radiation and chemotherapy treatments could enrich for CSCs [32]. The premise is that the treatments could upregulate the DNA repair mechanisms [33]. CSCs can self-renew, express stem cell-linked genes, and initiate tumors [34–36]. Unlike healthy stem cells, cancer progenitors can respond to the tissue microenvironment to dedifferentiation into CSCs [37, 38]. CSCs are mostly in cycling quiescence, can evade the immune response, and resistant treatment [20, 37, 39]. CSCs mostly express high levels of the multidrug resistance (MDR) genes [28, 40, 41]. CSCs can adapt cellular quiescence for long-term survival as dormant cells [21, 42].
In addition to the role of tissue microenvironment, CSCs, through autonomous mechanisms, maintain the epigenetic program to retain stemness [43–45]. Targeting of CSCs have proven to be a challenge due to tumor plasticity that causes the cancer cells to transition back and forth between progenitors and CSCs [46]. We report on distinct subsets of GBM cells using a lentivirus in which tandem repeats of Sox2 and Oct4a response element is linked to GFP [29]. We showed distinct GBM cell subsets. We also reported on another method in which tumorspheres can be passaged to select CSCs. The findings are discussed on the limitations of the method in the context of published studies. We also discussed how the finding could be applied with primary GBM cells.
Materials and Methods
Reagents, Antibodies, and Cytokines
Polybrene, recombinant epidermal growth factor (EGF), recombinant fibroblast growth factor basic (FGF) and B27 supplement were purchased from Fisher Scientific (Waltham, MA); Dulbecco’s Minimal Essential Media (DMEM), Eagle’s Minimum Essential Media (EMEM), fetal calf sera (FCS), trypsin, AldeRed Assay Kit, L-Glutamine and Penicillin-Streptomycin from Sigma-Aldrich (St Louis, MO); Mouse anti-human CD133-APC and mouse IgG-APC isotype from BD Bioscience (San Jose, CA)),
Cell Lines
Hek293T, T98G, A172 and U118 cells were purchased from American Type Culture Collection (ATCC, Manassas, Virginia). The cells were propagated as per manufacturer’s instructions. Cells were cultured in DMEM (A172, U118 and Hek293T) or EMEM (T98G) containing 10% FBS, 2 mM L-glutamine, 100 IU/ml penicillin, and 100 g/ml streptomycin. Cell culture media were changed every 2-3 days. At 80% confluency, cells were trypsinized and passaged.
Vectors
pOct4a-GFP was kindly provided by Dr. Wei Cui (Imperial College, UK) and was previously described [28]. SORE6 lentivirus was kindly provided by Dr. Wakefield from the National Cancer Institute (Bethesda, MD) [29]. The empty lentiviral vector (EV) was purchased from GeneCopeia (Rockville, MD).
Preparation of Lentiviral Particles
Lentiviral plasmids for SORE6-GFP was placed into One Shot Mach1T1 Phage- Resistant chemically competent E. Coli. DNA from the transformed bacteria was isolated with the Plasmid Miniprep Kit. The insert was verified by restriction digest with BamH1 and EcoRV followed by gel electrophoresis. The transformed bacteria were cultured and amplified in LB broth supplemented with ampicillin (50 g/ml). The plasmid DNA was collected from the transformed bacteria and then used to transduce HEK-293T cells. The DNA (5 g/ml) and lentiviral packaging plasmids (5 g/ml) were gently combined and then added to HEK-293T at 80-90% confluence. The plasmid mix was transferred to a tube containing 1 ml of Opti-MEM and gently mixed. TransIT-Lenti (Mirus Bio) reagent (30 l) was added and homogenized with the mixture. The sample was incubated at room temperature for 10 mins to allow the formation of transfection complexes. This was followed by drop wise addition to the HEK-293T cells. The cells were incubated at 37C. At 48 h, media were collected and centrifuged at 300 g for 10 mins to remove cellular debris. The supernatant containing the virus was filtered through a 0.45 m PVDF membrane and concentrated using the Lenti-X Concentrator. The viral solution was aliquoted in low-protein binding tubes at 10–10 IFU/ml and then stored at 80C.
Generation of SORE6 Stable Cell Lines
U118 and T98G cells were transduced with SORE6-GFP or Empty vector (EV)-GFP lentivirus at MOI of 5:1. The cells were cultured with complete media supplemented with 0.8 g/l of polybrene to facilitate transduction. After 48h, the cells were imaged under the EVOS Fl Auto 2 (ThermoFisher) for cellular fluorescence. Stable transduced cells were selected with 1–3 g/l of puromycin. The stable cell lines were maintained with 1 g/l puromycin for SORE6-GFP, and 2 g/l puromycin for cells with EV.
Generation of Stable T98G with pOct4a-GFP
T98G cells were transfected with pOct4a-GFP DNA using the lipofectamine 3000 transfection system. T98G cells were seeded at 70% confluency on 6-well plates. Confluence was achieved by overnight incubation. Opti-MEM medium (125 l) was mixed with 5 l of Lipofectamine 3000 reagent into a tube followed by vortexing. Into a second tube, 5 l of P3000 reagent and 2.5 g of the plasmid DNA was added followed by thorough mixing. The tubes were incubated for 10 min at room temperature. The components were added dropwise to the cells and transfection was allowed by incubation for 2–3 days. The transfected cells were selected with 200–300 g/ml of G418. Clones were selected and expanded to generate a stable cell line with pOct4a-GFP.
Real Time PCR
Total RNA was isolated with TRIzol reagent according to the manufacturer’s instructions. The RNA was reverse transcribed into cDNA with the High-Capacity cDNA Reverse Transcription kit and then amplified using the GeneAmp PCR System 9700. The cDNA was diluted to 200 ng/l and then mixed with SYBR Green PCR Master Mix, primers of interest, and deionized water. Real time PCR was conducted on a 7300 Real-time PCR system (ThermoFisher Scientific) with cycle conditions of 50C for 2 mins, 95C for 10 mins, followed by 40 cycles of 95C for 15 secs and 60C for 1 min. The following primers were used on the PCR mix: Oct4a: Forward 5 act atg cac aac gag agg at 3, Reverse 5 tac agt gca gtg aag tga gg 3; Sox2: Forward 5 agg ata agt aca cgc tgc cc 3; Reverse 5 taa ctg tcc atg cgc tgg tt 3; Nanog: Forward 5 acc caa tcc tgg aac aat 3, Reverse 5 cac tgg cag gag aat ttg 3; KLF4: Forward 5 aac ctt acc act gtg act gg 3, Reverse 5 cat atc cac tgt ctg gga tt 3; Notch1: Forward 5 cca agt ata gcc tat ggc aga a 3, Reverse 5 aag tct gac gtc cct cac tg 3; GAPDH: Forward 5 cag aag act gtg gat ggc c 3, Reverse: 5 cca cct tct tga tgt cat c 3.
Fluorescence Assisted Cell Sorting (FACS) of SORE6-GFP GBM Cells
T98G and U118 SORE-6-GFP cells were sorted into GFP-Hi, med, lo and negative cells using Fluorescence assisted cell sorting (FACS). Cells (10) were washed with 1X PBS and then filtered to eliminate cell clumps. The latter was accomplished by adding the cells to Falcon Round-Bottom Polystyrene test tubes (Fisher Scientific). Sorting used flow tubes containing HEPES buffer and 2% FBS. Sorting gated cells with relative GFP fluorescence. The cells were collected into tubes coated with 4% FBS. The sorted cells were collected by centrifugation at 300 g for 10 min followed by resuspension in test media.
Tumorsphere Assay
Tumorsphere assay for U118 and T98G -SORE6 cells was performed as described [28]. Briefly, cells were sorted and then added at 1–4 cells/well into 96-well low attachment plates (Costar, Corning, NY). The culture media contained Neurobasal media, 2% B27, 20 ng/nl FGF and 20 ng/ml EGF. Spheres were dissociated using mechanical disruption and serially diluted into a new low attachment 96-well plates. The process was repeated for 3 passages.
In other studies, heterogenous T98G and U118 cells were seeded at density of 10 cells into low attachment 6-well plates, and cultured using Neurobasal media supplemented with B27, FGF and EGF, until spheres were observed. Half the population was collected and stored in trizol for real time PCR, and the rest were mechanically dissociated and serially diluted and replated into fresh plates. The process was repeated up to passage 5.
ALDH Assay
The ALDH assay was performed with 10 U118 and T98G cells in 1 mL of AldeRed Assay Buffer. 5 L of AldeRed Reagent was added to the cells. Half of the cell solution was added to a new tube with 5 L of DEAB, a Aldehyde Dehydrogenase 1 inhibitor, serving as a control. Both tubes were incubated for 30 minutes at 37C. This was followed by washing, and resuspension in PBS. The cells were imaged on a Becton Dickinson FACS Caliber, capturing on Filter2 (585/42nm).
In vivo Passage of Tumor
Female athymic nude mice, 6 wks, were purchased from Charles River (Fairfield, NJ). The use of mice was approved by Rutgers Institutional Animal Care and Use Committee, Newark, NJ. Mice were housed at an AAALAC accredited facility.
Mice were injected subcutaneously with 10 T98G or U118-GFP -Hi cells in 200 l of PBS, mixed with equal amount of Matrigel. The cells were injected subcutaneously on the mice flanks and at the back. The tumors were excised and then digested with collagenase. The clumps were filtered with sterile cell straining with 100 M Nylon Mesh. the suspension tells collected and the process repeated as before for secondary tumor formation.
Statistical Analysis
Data was analyzed using student’s t-test and ANOVA to compare between groups. A p-value of 0.05 was considered significant.
Results
Establishing GBM-SORE6-GFP Cells
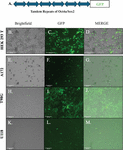
Six tandem repeats of SOX2-Oct4a-response elements (SORE6), inserted in a lentiviral reporter system controlled the expression of GFP [29] (Figure 1A). Cells expressing the lentivirus will respond to stem cells with increased levels of Sox2 and Oct4a proteins, which will be reflected by GFP intensity [29]. To this end, we used the lentiviral vector to demarcate GBM cell subsets into a working hierarchy. We adapted the methods with breast cancer cells that selected subsets with a plasmid in which GFP was under the regulation of the full length Oct4a promoter [28, 47].
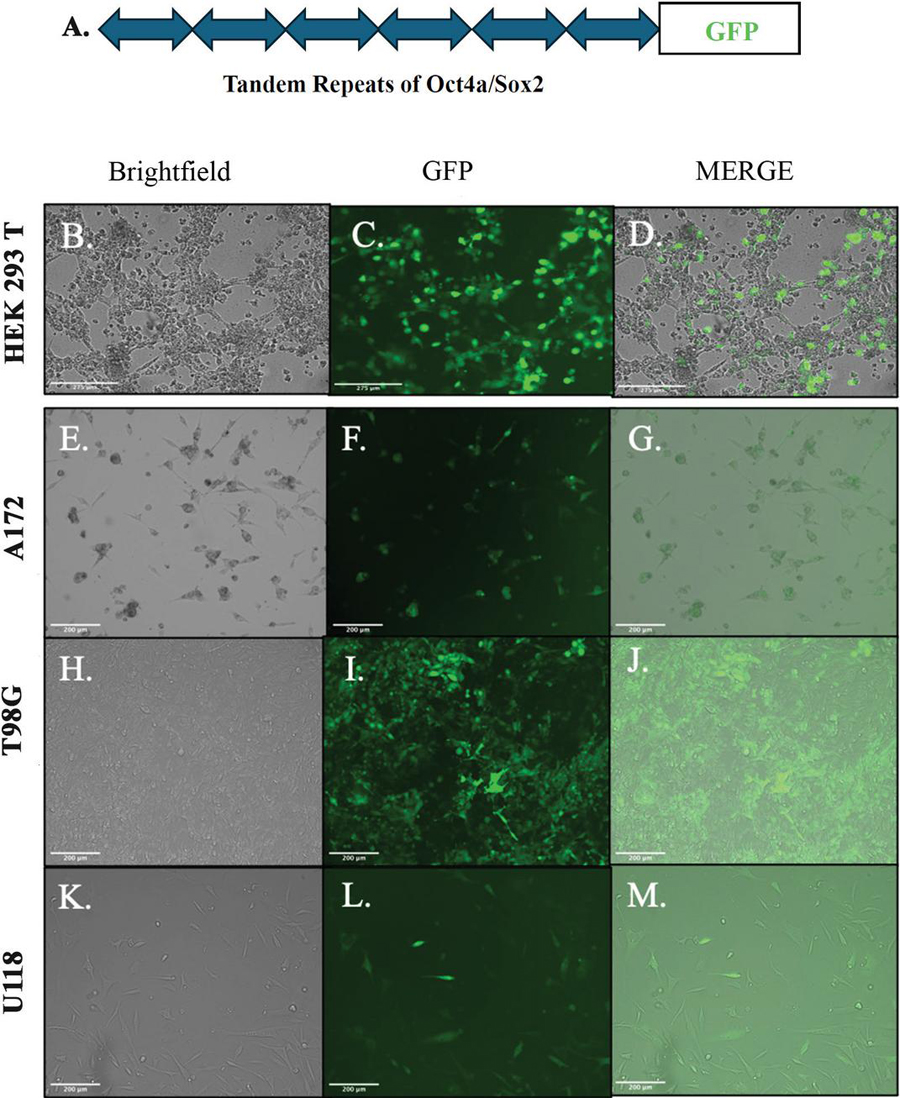
Figure 1 Establishing stable GBM cell lines with SORE6-GFP. A) Shown is the basic structure of the stem cell repeats that regulate the expression of GFP. B-D) SORE6 vector in the propagating HEK293 cell line. E-M) Stable transduction of A172, T98G and U118 cells with SORE6-GFP lentivirus.
The SORE6-GFP lentivirus was generated with transduced HEK293 T cells and the viral particles released into the media were quantified by ELISA. We obtained particles, 10–10 Infection units/ml (IFU/ml). U118, T98G and A172 GBM cells were transduced with the lentivirus using an MOI of 5:1. After 48 h, the cells were analyzed for fluorescence by flow cytometry and imaging with the EVOS Fl2. Stable transfectants were selected and maintained with puromycin. Three stable cell lines, shown in Figure 1B, were used in the studies described in this report.
Working GBM Cell Hierarchy
We used flow cytometry to analyze the scatter plots of U118 and T98G cells for relative fluorescence (GFP). The cell populations were assigned into a hierarchy, based on relative fluorescence intensities. The subsets were assigned as GFP-High (Hi), -medium (Med), -Low or -Negative (Neg) (Figures 2A and 2B). Similar pattern of fluorescence intensities were noted for both cell lines. Since fluorescence intensities have been shown to be proportional to the levels of stem cell expressed genes, we established a hierarchy based on relative fluorescence intensities (Figure 2C).
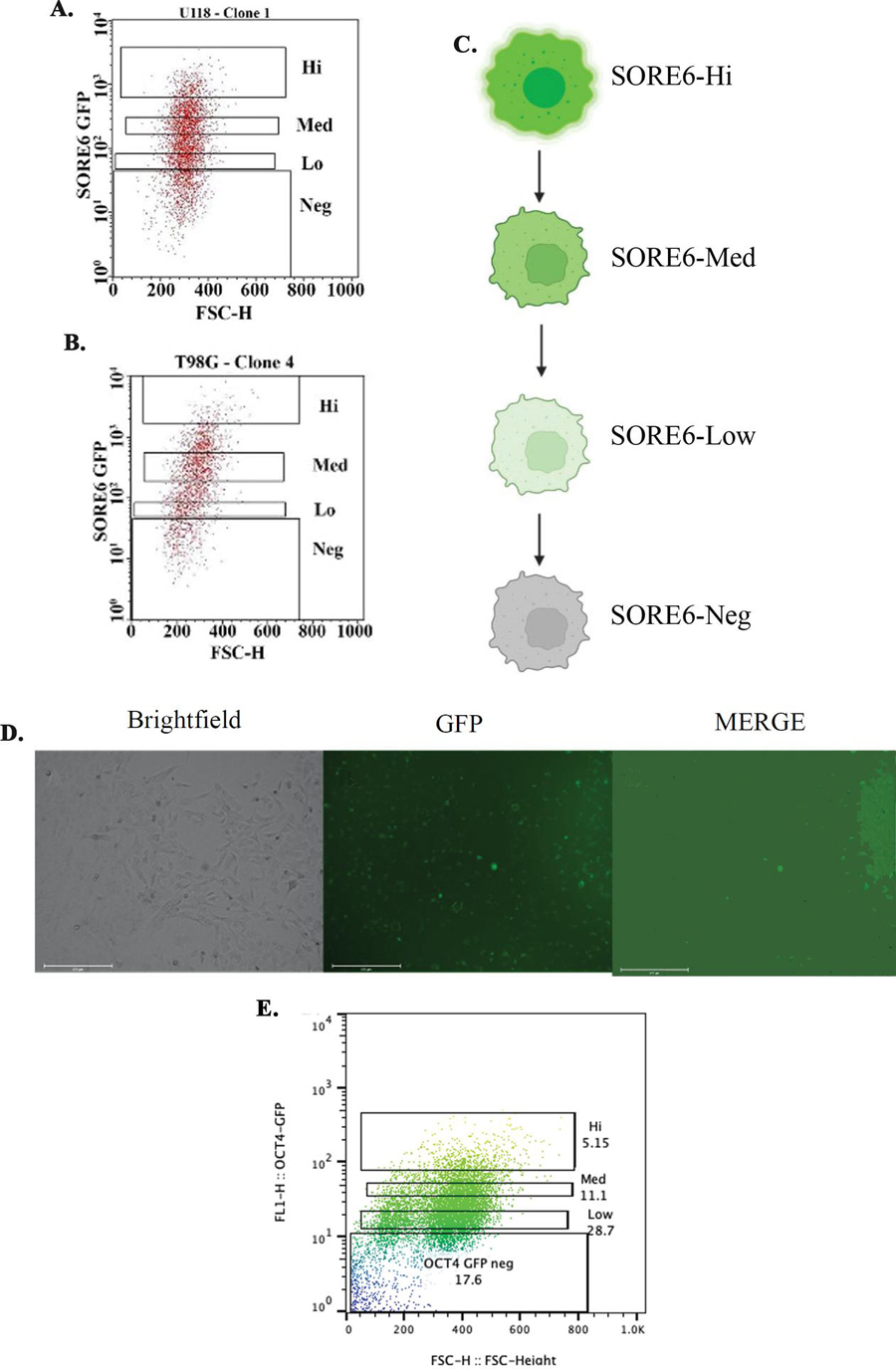
Figure 2 Scatter plot of GBM cells with stable SORE6-GFP. A & B) Shown are representative scatter plots for U118 (A) and T98G (B) to delineate fluorescence as High (Hi), medium (med), low (lo) and negative (neg). C) The assigned boxed sections in A and B were demarcated in a working hierarchy. D) Shown is representative transfection of T98G cells with pOct4a-GFP. E) Representative scatter plot of the transfectants from ‘D’ showing cell subsets, based on relative fluorescence intensities.
We next analyzed T98G cells to determine if we can achieve similar hierarchy with the full Oct4a promoter linked to GFP (pOct4a-GFP). Similar to the studies with breast cancer, we stably transfected T98G with pOct4a-GFP [28] (Figure 2D). Although we noted distinct population based on relative fluorescence intensities, the demarcation was not as widespread as the scatter plot obtained with the lentivirus (Figure 2C versus, Figures 2A and 2B). Together, these findings indicated that the SORE6 lentivirus could serve as a key experimental method to advance studies with GBM cell subsets.
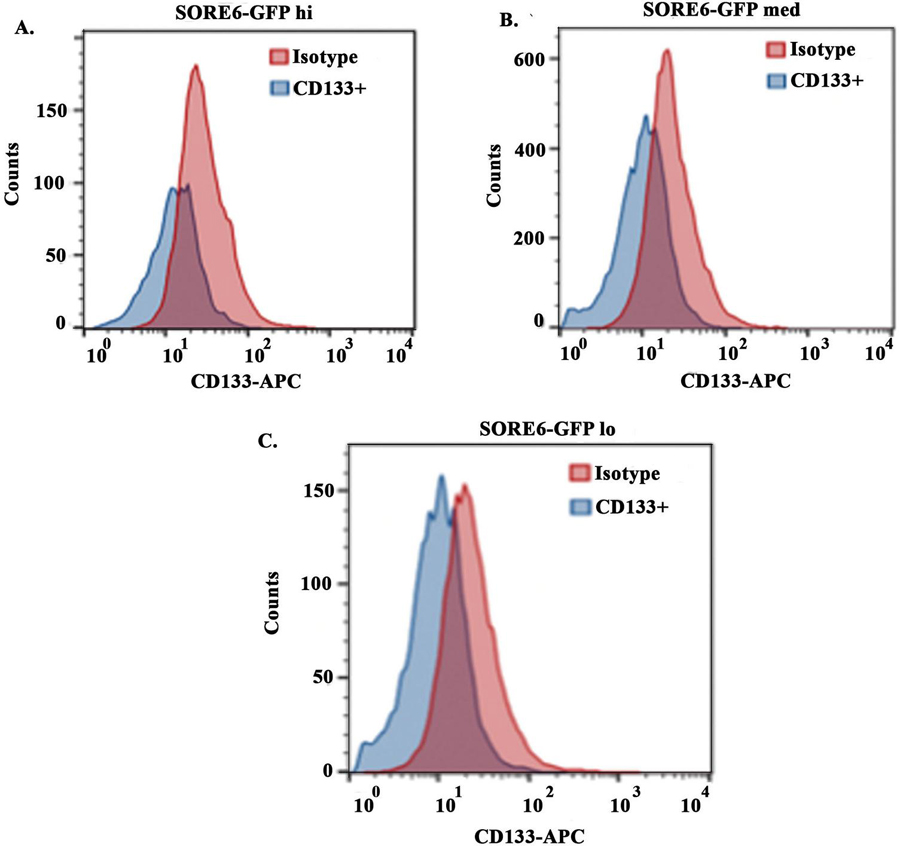
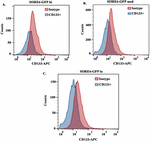
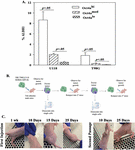
Figure 3 Assessing CD133+ on SORE6 GBM subsets. Flow cytometry for CD133+ was evaluated within SORE6-GFP expressing GBM subsets. Isotype control represent SORE-6-GFP hi (A), med (B) and lo (C). The results are a mean of three different independent experiments.
CD133 on SORE6 Cells
Since CD133 has been reported on GBM CSCs [48], we analyzed the SORE6 GBM subsets from T98G for CD133 by flow cytometry. The histogram labeled as isotype represents the SORE6-GFP subsets (Figure 3). The mean fluorescence intensity for CD133 with each GBM subset was similar (Figure 3). The intensities were consistently lower than the baseline GFP, which is label as isotype. These results indicated that SORE6-GFP cells expressed CD133 on each of the three GBM subsets. This indicated that CD133, as a marker of CSCs, would be better if combined with other methods to define GBM cell subsets.
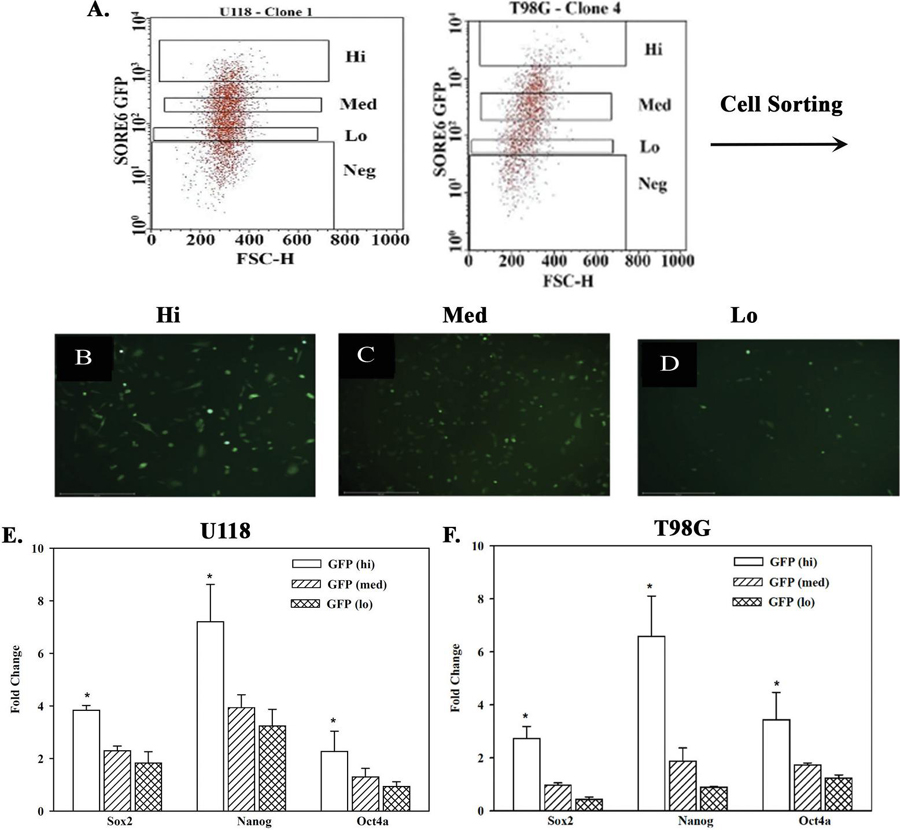
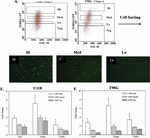
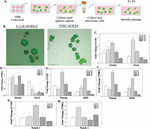
Figure 4 Real time PCR for stem cell-linked genes in GBM subsets. A) Representative scatter plot of SORE6-GFP U118 and T98G cells to depict how each subset was selected for sorting. B-D) Representative images of sorted SORE6-GFP Hi, med, Lo cells. E and F) Real time PCR for Sox2, Nanog and Oct4a with RNA from GFP-Hi, -med and -lo sorted from U118 (E) or T98G SORE6 cells. The results are presented as the meanSD of three biological replicates.
Real Time PCR for Stem Cell-linked Genes in SORE6-GFP GBM Subsets
In this set of studies, we asked if the working hierarchy shown in Figure 2C correlated with the expression of three stem cell genes, Sox2, Nanog and Oct4a. We sorted subsets of GBM cells from SORE6-GFP U118 and T98G cells – hi, med and low (Figures 4A–4D). The method of sorting was previously described for breast cancer cells and was adapted for this study [47]. The relative GFP intensities of the sorted cell subsets are shown as microscopic fluorescence images (Figures 4B–D). Real time PCR for Sox2, Nanog and Oct4a showed similar results for both U118 and T98G (Figures 4E and 4F). Each transcript was significantly (p 0.05) increased in the hi subsets as compared to med and lo subsets. These findings supported the working hierarchy shown in Figure 2C with respect to relative cell maturity.
Tumorsphere Formation by Subsets of GBM SORE6-GFP
We compared sorted U118-SORE6-GFP subsets during serial cell passaging for tumorsphere formation. Cells were added at 4 cells/well in low attachment 96-well plates with weekly replacement of media (Figure 5A). At each passage, the number of spheres were counted and then mechanically dissociated for passaging up to three times (Figure 5A). Parallel studies were performed with CD133+ cells, sorted from the SORE6-GFP GBM cells. Representative spheres during passages are shown for each cell subset (Figure S1). Only SORE6-GFP hi subset showed spheres up to passage 3 (Figure 5B). The hi subset showed increased number of tumorspheres at passaging (Figure 5B). We surmised that at each passage, there were multiple stem cells within the sphere that accounted for the increase in total spheres. In contrast to the hi subset, the number of spheres for the medium and low subsets were low until passage 3 (Figure 5B). We conducted similar studies with spheres from CD133+ cells that were sorted from U118 SORE6-GFM cells. We noted significant (p 0.05) increases in the number of spheres with each passage (Figure 5C). Although increased, passage 3 spheres were significantly less than the number of tumorspheres at passage 2 (Figure 5C).
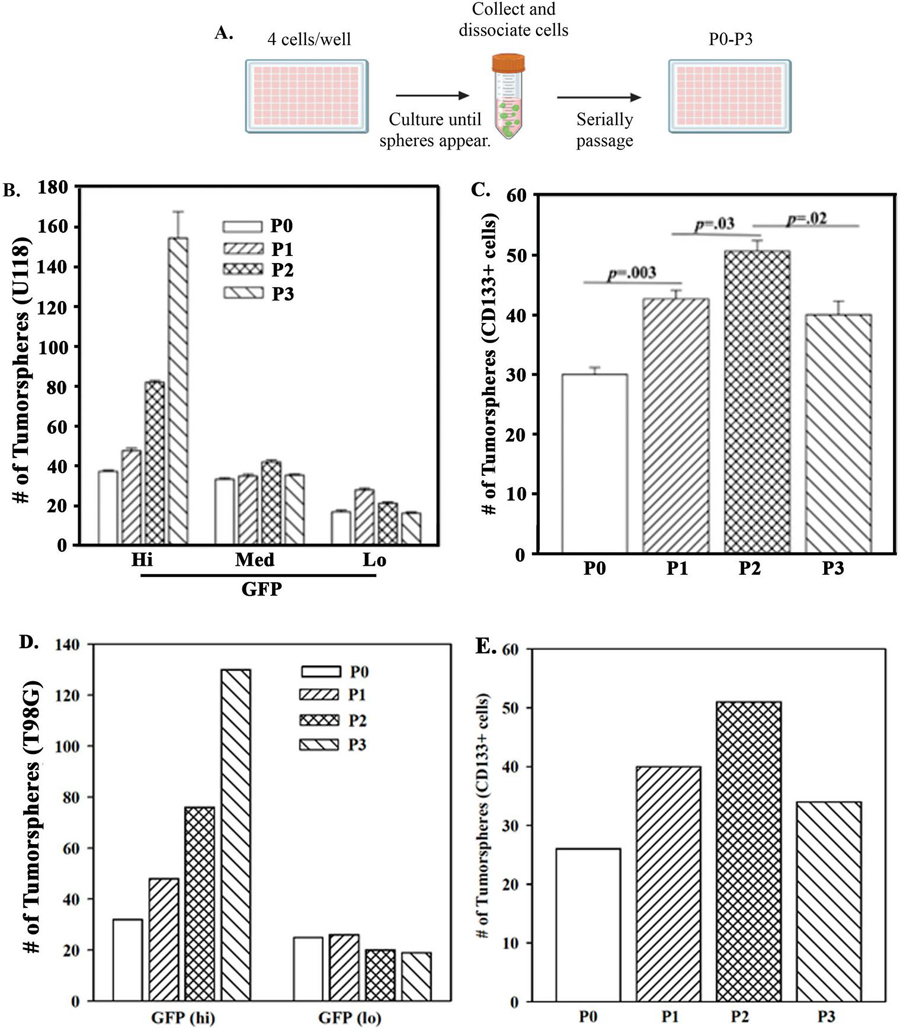
Figure 5 Tumorspheres from serially passaged SORE6-GFP subsets. A)Diagram depicts the approach using SORE6-GFP GBM cells (U118 and T98G). B) Quantification of tumorspheres from U118-SORE6-GFP Hi, Med, Low were taken from the passaged spheres (Figure S1); C) Tumorspheres obtained with sorted CD133+ cells at each passage. The results are presented as the meanSD of three biological replicates. D) Quantification of tumorspheres from T98G-SORE6-GFP Hi and lo in two biological replicates. E) The studies in ‘C’ was repeated with two biological replicates.
Studies with SORE6-GFP-T98G indicated similar findings as shown for U118 with respect to sphere formation. However, unlike the studies with U118, we used two subsets – hi and lo in two biological replicates (Figure S2). Passaging increased the number of spheres with T98G GFP-hi cells whereas the low number of spheres for the lo subsets remained constant (Figure 5D). Parallel studies with SORE6-GFP T98G CD133+ cells showed an increase in tumorsphere (Figure 5E). Similar to the drop in spheres at passage 3 for U118 (Figure 5C), we also noted a similar decrease for T98G at passage 3 (Figure 5E). Thus, SORE6-GFP cells could be a model to be incorporated in isolating tumor cells with the incorporation of CD133 marker.
ALDH 1 Activity in GBM Cell Subsets and In vivo Passaging
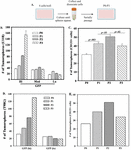
The formation of tumorspheres were insufficient to assign the SORE6-GFP hi cells as CSCs (Figure 5). CSCs have been reported to express the highest level of ALDH 1 [26, 28]. We therefore asked if SORE6-GFP hi GBM cells express ALDH activity. We assessed U118 and T98G SORE6-GFP cells for ALDH 1 in the Hi, med and lo subsets. The highest percentage of ALDH positive cells was observed in the GFP-Hi subset with the least in the lo subset (Figure 6A).
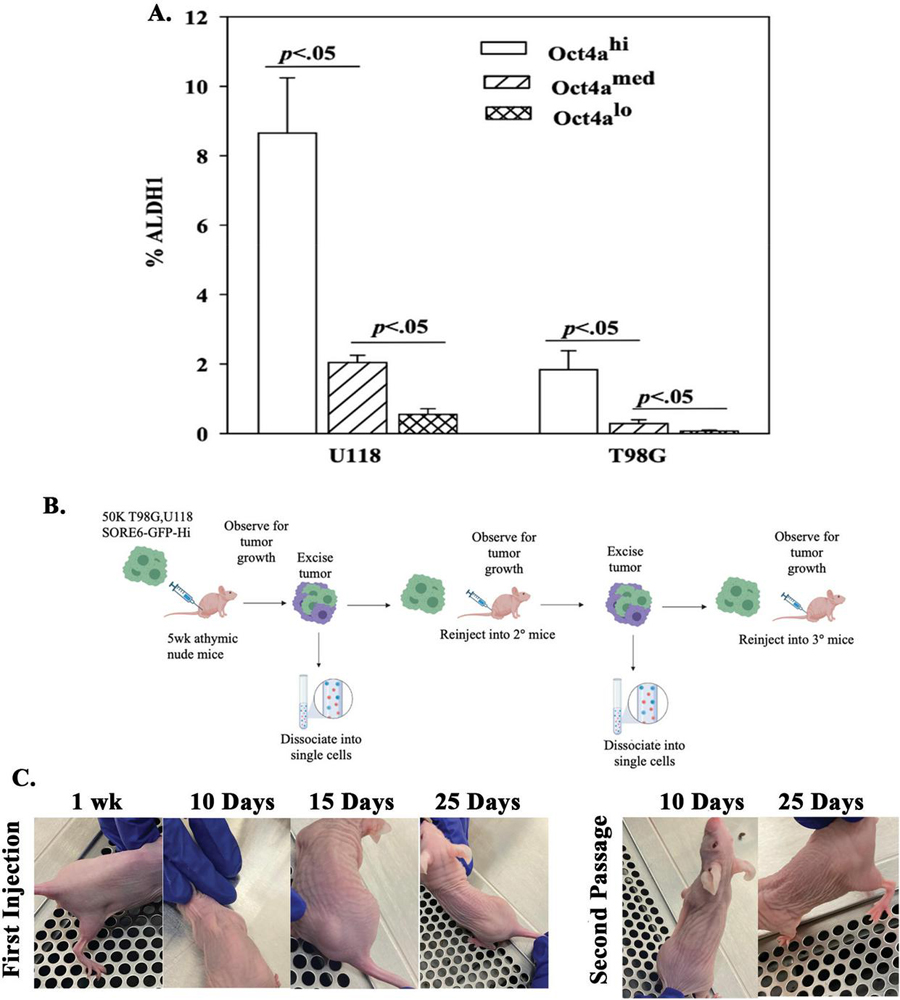
Figure 6 ALDH assay with subsets of T98G-SORE6-GFP. A) The values from Figure S3 are plotted as the meanSD of three biological replicates. B) Shown is the protocol used to inject 5x10 cells injected subcutaneously into each flank and back of nude mice. C) Shown are representative images of tumor growth from primary injections from Day 7 up to Day 25, and secondary injections from cells isolated from excision of primary tumor at Days 10 and 25.
Thus far, the in vitro studies suggested that SORE6-GFP hi cells could be CSCs. We next determined if these cells could be passaged in vivo using athymic female nude mice. U118 and T98G SORE6-GFP-hi cells cells were injected subcutaneously into each flank and at the back of nude mice (Figure 6A). The mice were observed for tumor growth up to day 30. The primary tumors were excised, digested and filtered into cell suspension. The hi subsets were successful with respect to serial passaging in another group of mice (Figure 6C). The in vivo studies supported SORE6-GFP-hi cells as CSCs.
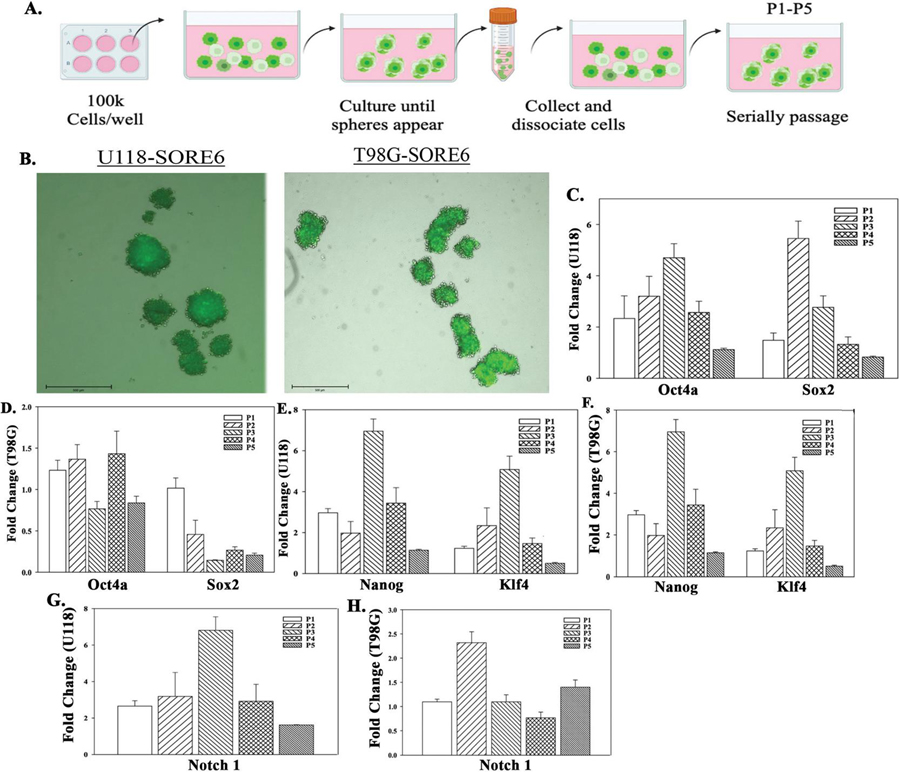
Figure 7 Tumorsphere formation in low attachment plates and heterogenous GBM cells. A) Diagram showing the protocol used to passage spheres formed by heterogeneous GBM cells. The diagram was created with BioRender.com. B) Representative spheres from U118-SORE6-GFP and T98G-SORE6-GFP cells at 10X magnification. C-H) Real time PCR using RNA from the spheres at passages (P) 1-5. Stem cell genes – Oct4a and SOX2 (C and D), nanog and Klf4, and (E and F), and Notch1(G and H). The results are presented as the meanSD of three biological replicates.
CSC Selection by Sphere Passaging from Heterogeneous GBM Cells
The process of isolating CSCs continues to prove that even cells that are functionally CSCs are heterogeneous [47]. CSCs have been reported from passaging of ovarian cancer cells [49]. We applied a similar method with unsorted/heterogeneous GBM cells (Figure 7A). Thus, SORE6-GFP U118 and T98G cells were cultured in low attachment 6-well plates supplemented with Neurobasal media until sphere-like structures developed (Figure 7B). Half the spheres were collected after each passage for PCR and the other half, dissociated, diluted and plated into fresh plates. This process was repeated up to 5 passages.
Real time PCR for stem cell markers, Oct4a, Sox2, Nanog, Klf-4 and Notch1, showed increases in the levels of these genes at passaging (Figures 7C–7G). The values peaked at passage (P) 2 or P3 followed by decrease at P4 and P5.
Discussion
This study describes several methods to develop a hierarchy of GBM cells. Studies comparing a plasmid and lentiviral system determined that the latter was more efficient with respect to distinct separation of the various subsets (Figure 2). Both methods used cells with stable transfectants of vectors linked to the GFP reporter gene. The plasmid consisted of the full-length Oct4a promoter, and the lentivirus with tandem repeats of Sox2 and Oct4a interacting sites (SORE6-GFP) [28, 29]. The lentiviral system could enhance detecting two stem cell genes whereas use of the plasmid will activate GFP with high levels of Oct4a. Theoretically, there is an advantage to the Oct4a full regulatory region since other stem cell-linked transcription factors within CSCs could activate the 5 regulatory region. However, in this study, SORE6-GFP was more efficient to demarcate distinct GBM subsets.
This study clarified that isolation of GBM cell subsets requires multiple methods that include phenotype and function. The working hierarchy that we developed is mostly based on relative GFP, but was supported by stem cell gene expressions and other functional studies (Figures 2C–7). The relative cell maturity was investigated by real time PCR for stem cell genes, which showed direct relationship to GFP intensity (Figure 4).
The literature on CD133 established that this membrane protein could be a marker of CSCs, including those within GBM. However, the literature suggested inconsistency of CD133 as a solid marker of CSCs [25, 48]. In our studies, we showed its expression on each of the three GBM subsets (Figure 3). Despite its presence on each subset, sorting of all CD133+ cells within GBM cells indicated higher number of tumorspheres with passaging (Figure 5). These findings suggested that CD133+ cells could be a key marker to define GBM CSCs but demonstrated that a single marker would not be adequate to identify CSCs. In this case, CD133 selection should be used in conjunction with other methods. There is however caution for selection of cells from primary GBM tumor because CD133 is expressed on neural cells [48]. A supported method is described in Figure 7 in which the GBM cells could eliminate the use of reporter gene system and to select CSCs by serial passaging of spheres.
The issue with using a reporter gene system is the difficulty of demarcating primary GBM cells since selection would change the primary tumor and the cell types. Serial passing of spheres with heterogeneous tumor cells in low attachment plates leads to the selection of cells that are more likely CSCs (Figure 7). This method is exciting since it could be applied to primary tumor cells. Further studies are needed to determine if the spheres were similar to those isolated with SORE6-GFP hi GBM cells. We propose that omics studies with these spheres, including single cell sequencing, could be insightful for additional markers to isolate primary CSCs.
The tumorsphere assay with GBM subsets added 1–4 cells/well isolated from U118 and T98G cells (Figure 5). The results indicated the highest number of tumorspheres within SORE6-GFP-hi cells. However, the total numbers increased with passaging. Perhaps, this is due to some wells containing 4 cells, when passaged, will lead to increased numbers of SORE6-GFP-Hi spheres. Regardless, it is clear that only the GFP-hi subsets were able to show timeline increases in tumorspheres. These findings correlated with increased levels of ALDH 1, which adds to the support of the CSC identity. However, similar to the discussion above, ALDH 1 could be expressed in stem and progenitor cells, further supporting the use of multiple methods [50]. This study examined each subset of GBM cells for ALDH 1. High ALDH 1 was noted to be high in the SORE6-GFH hi cells and this gradually decreased with reduced GFP (Figures S3 and 6A). However, the efficiency was quite low perhaps due to the high expression of ALDH1A1 in GBM cells, particularly recurrent GBM cells [50, 51] and might not completely delineate the heterogeneity of CSCs. Thus, the SORE6-GFP method might be more efficient and can be used either individually or in tandem with ALDH 1 assay. Finally, athymic nude mice were injected subcutaneously with SORE6-GFP-Hi cells and observed for tumor growth for about 30 days. The tumor was excised, and the tissue digested and serially diluted and reinjected into secondary mice and again observed for tumor growth. I again confirmed tumor growth at the injection site (Figure 6B), confirming the tumor initiating ability of SORE6-GFP-Hi GBM cells.
Overall, the SORE6-GFP reporter system could be an effective method to efficiently identify and isolate CSCs from U118 and T98G GBM cells, with the GFP-Hi subset as the long-term CSCs with tumor initiating properties. The SORE6-GFP reporter system also recapitulates the CSC heterogeneity and can be used to further study different CSC subtypes. However, this study clearly indicates that the identification of CSCs requires multiple types of assays to identify CSC [28]. This study will allow for indepth study to treat GBM, which is the most aggressive brain tumors with poor prognosis [52]. The method used to select CSCs from heterogeneous GBM cells could be relevant to primary tissues and needs further pursuit (Figure 7).
Acknowledgement
This work is in partial fulfillment for VHS doctoral thesis.
Authors’ Contributions
VHS wrote the first draft of the manuscript, conducted the experiments and analyzed the data; AS, AP and JB-S edited the manuscript and conducted the experiments; MG contributed to establishing the vector systems used in the manuscript and edited the manuscript; AH contributed to the concept and edited the manuscript; PR supervised and conceptualized the study, approved the final manuscript.
References
[1] Hanif F, Muzaffar K, Perveen K, Malhi SM, Simjee Sh U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac J Cancer Prev. 2017;18(1):3–9.
[2] Alifieris C, Trafalis DT. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82.
[3] Grech N, Dalli T, Mizzi S, Meilak L, Calleja N, Zrinzo A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus. 2020;12(5):e8195.
[4] Mathur R, Wang Q, Schupp PG, Nikolic A, Hilz S, Hong C, et al. Glioblastoma evolution and heterogeneity from a 3D whole-tumor perspective. Cell. 2024;187(2):446–63:e16.
[5] Alexander BM, Cloughesy TF. Adult Glioblastoma. J Clin Oncol. 2017;35(21):2402–9.
[6] Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231–51.
[7] Armstrong TS, Bishof AM, Brown PD, Klein M, Taphoorn MJ, Theodore-Oklota C. Determining priority signs and symptoms for use as clinical outcomes assessments in trials including patients with malignant gliomas: Panel 1 Report. Neuro Oncol. 2016;18 Suppl 2(Suppl 2):ii1–ii12.
[8] Hooper GW, Ginat DT. MRI radiomics and potential applications to glioblastoma. Front Oncol. 2023;13:1134109.
[9] Nam JY, de Groot JF. Treatment of Glioblastoma. J Oncol Pract. 2017;13(10):629–38.
[10] Mohammed S, Dinesan M, Ajayakumar T. Survival and quality of life analysis in glioblastoma multiforme with adjuvant chemoradiotherapy: a retrospective study. Rep Pract Oncol Radiother. 2022;27(6):1026–36.
[11] Singh N, Miner A, Hennis L, Mittal S. Mechanisms of temozolomide resistance in glioblastoma – a comprehensive review. Cancer Drug Resist. 2021;4(1):17–43.
[12] Ahmed MH, Canney M, Carpentier A, Idbaih A. Overcoming the blood brain barrier in glioblastoma: Status and future perspective. Rev Neurol. 2023;179(5):430–6.
[13] Abe H, Natsumeda M, Okada M, Watanabe J, Tsukamoto Y, Kanemaru Y, et al. MGMT Expression Contributes to Temozolomide Resistance in H3K27M-Mutant Diffuse Midline Gliomas. Front Oncol. 2019;9:1568.
[14] Munoz JL, Rodriguez-Cruz V, Ramkissoon SH, Ligon KL, Greco SJ, Rameshwar P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget. 2015;6(2):1190–201.
[15] Bausart M, Preat V, Malfanti A. Immunotherapy for glioblastoma: the promise of combination strategies. J Exp Clin Cancer Res. 2022;41(1):35.
[16] Hill SA, Blaeser AS, Coley AA, Xie Y, Shepard KA, Harwell CC, et al. Sonic hedgehog signaling in astrocytes mediates cell type-specific synaptic organization. Elife. 2019;8.
[17] Chandra V, Das T, Gulati P, Biswas NK, Rote S, Chatterjee U, et al. Hedgehog signaling pathway is active in GBM with GLI1 mRNA expression showing a single continuous distribution rather than discrete high/low clusters. PLoS One. 2015;10(3):e0116390.
[18] Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull. 2017;7(3):339–48.
[19] Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–17.
[20] Lei MML, Lee TKW. Cancer Stem Cells: Emerging Key Players in Immune Evasion of Cancers. Front Cell Dev Biol. 2021;9:692940.
[21] Talukdar S, Bhoopathi P, Emdad L, Das S, Sarkar D, Fisher PB. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv Cancer Res. 2019;141:43–84.
[22] Wei Y, Li Y, Chen Y, Liu P, Huang S, Zhang Y, et al. ALDH1: A potential therapeutic target for cancer stem cells in solid tumors. Front Oncol. 2022;12:1026278.
[23] Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W, Kopetz S, et al. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One. 2011;6(6):e20636.
[24] Ren F, Sheng WQ, Du X. CD133: a cancer stem cells marker, is used in colorectal cancers. World J Gastroenterol. 2013;19(17):2603–11.
[25] Glumac PM, LeBeau AM. The role of CD133 in cancer: a concise review. Clin Transl Med. 2018;7(1):18.
[26] Rodriguez-Torres M, Allan AL. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin Exp Metastasis. 2016;33(1):97–113.
[27] Zanoni M, Bravaccini S, Fabbri F, Arienti C. Emerging Roles of Aldehyde Dehydrogenase Isoforms in Anti-cancer Therapy Resistance. Front Med (Lausanne). 2022;9:795762.
[28] Patel SA, Ramkissoon SH, Bryan M, Pliner LF, Dontu G, Patel PS, et al. Delineation of breast cancer cell hierarchy identifies the subset responsible for dormancy. Sci Rep. 2012;2:906.
[29] Tang B, Raviv A, Esposito D, Flanders KC, Daniel C, Nghiem BT, et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Reports. 2015;4(1):155–69.
[30] Ivanova A, Kravchenko D, Chumakov S. a Modified Lentivirus-Based Reporter for Magnetic Separation of Cancer Stem Cells. Molecular Biology. 2020;54:82–8.
[31] Ribeiro IG. Isolation and Characterization of Cancer Stem Cells (CSCs) from Colorectal Cancer-Transcription Factors Involved in Their Reprogramming. Universidade do Porto (Portugal); 2020.
[32] Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Seminars in cancer biology, 2015. Elsevier: 28–35.
[33] Liu Q, Guo Z, Li G, Zhang Y, Liu X, Li B, et al. Cancer stem cells and their niche in cancer progression and therapy. Cancer Cell Int. 2023;23(1):305.
[34] Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44(12):2144–51.
[35] Swain N, Thakur M, Pathak J, Swain B. SOX2, OCT4 and NANOG: The core embryonic stem cell pluripotency regulators in oral carcinogenesis. J Oral Maxillofac Pathol. 2020;24(2):368–73.
[36] Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–91.
[37] Chen K, Zhang C, Ling S, Wei R, Wang J, Xu X. The metabolic flexibility of quiescent CSC: implications for chemotherapy resistance. Cell Death Dis. 2021;12(9):835.
[38] Sandiford OA, Donnelly RJ, El-Far MH, Burgmeyer LM, Sinha G, Pamarthi SH, et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021;81(6):1567–82.
[39] de Morree A, Rando TA. Regulation of adult stem cell quiescence and its functions in the maintenance of tissue integrity. Nat Rev Mol Cell Biol. 2023;24(5):334–54.
[40] Choi CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005;5:30.
[41] Vera-Ramirez L, Hunter KW. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Res. 2017;6:2134.
[42] Endo H, Inoue M. Dormancy in cancer. Cancer Sci. 2019;110(2): 474–80.
[43] Ferrer-Diaz AI, Sinha G, Petryna A, Gonzalez-Bermejo R, Kenfack Y, Adetayo O, et al. Revealing role of epigenetic modifiers and DNA oxidation in cell-autonomous regulation of Cancer stem cells. Cell Commun Signal. 2024;22(1):119.
[44] Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest. 2010;120(6):2030–9.
[45] Luo M, Li JF, Yang Q, Zhang K, Wang ZW, Zheng S, et al. Stem cell quiescence and its clinical relevance. World J Stem Cells. 2020;12(11):1307–26.
[46] Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017;50(3):117–25.
[47] Bliss SA, Paul S, Pobiarzyn PW, Ayer S, Sinha G, Pant S, et al. Evaluation of a developmental hierarchy for breast cancer cells to assess risk-based patient selection for targeted treatment. Sci Rep. 2018;8(1):367.
[48] Ahmed SI, Javed G, Laghari AA, Bareeqa SB, Farrukh S, Zahid S, et al. CD133 Expression in Glioblastoma Multiforme: A Literature Review. Cureus. 2018;10(10):e3439.
[49] Malekshah OM, Sarkar S, Nomani A, Patel N, Javidian P, Goedken M, et al. Bioengineered adipose-derived stem cells for targeted enzyme-prodrug therapy of ovarian cancer intraperitoneal metastasis. J Control Release. 2019;311–312:273–87.
[50] Prasmickaite L, Engesaeter BO, Skrbo N, Hellenes T, Kristian A, Oliver NK, et al. Aldehyde dehydrogenase (ALDH) activity does not select for cells with enhanced aggressive properties in malignant melanoma. PLoS One. 2010;5(5):e10731.
[51] Clark DW, Palle K. Aldehyde dehydrogenases in cancer stem cells: potential as therapeutic targets. Ann Transl Med. 2016;4(24):518.
[52] Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci U S A. 2000;97(12):6242–4.
International Journal of Translational Science, Vol. 1, 177–200.
doi: 10.13052/ijts2246-8765.2024.033
© 2024 River Publishers