Breast Cancer Dormancy – Lessons Learnt
Jiaxing Yang†, Hana Choi†, Erikka Chowdhury and Jean-Pierre Etchegaray*
Rutgers, The State University of New Jersey, School of Arts and Sciences – Newark, Department of Biological Sciences, New Jersey, United States
E-mail: etchegje@newark.rutgers.edu
*Corresponding Author
†Co-first authors
Received 15 October 2024; Accepted 21 October 2024
Abstract
Despite early diagnosis and improved treatments, breast cancer remains a challenging disease. A strategic advantage for breast cancer recurrence is the ability of breast cancer cells to become quiescent and survive for decades in a dormant state within the endosteal region of the bone marrow. Breast cancer dormancy is triggered by exosome vesicles secreted by mesenchymal stem cells residing in the bone marrow. By mechanisms yet to be determined, dormant breast cancer cells are awakened leading to resurgence and metastasis. Experimental evidence supports the notion that dormant breast cancer cells are cancer stem cells recognized as tumor initiating and propagating cells with chemoresistant and metastatic properties. These cells represent less than 2% of the total tumor mass, which impose a significant barrier for their therapeutic targeting. This review focuses on cellular and molecular properties of breast cancer dormancy including tumor microenvironment, epigenetic regulation, cell signaling and metabolic reprogramming.
Keywords: Breast cancer, dormancy, metastasis, bone microenvironment, bone marrow.
Introduction
Breast cancer (BC) is a heterogeneous disease that can be categorized into three groups based on molecular and histological properties: (1) hormone receptors, including estrogen receptor (ER) or progesterone receptor (PR) expressed BC, (2) human epidermal growth factor 2 (HER2) expressed BC, (3) triple-negative breast cancer (TNBC) (ER, PR, HER2) [1]. TNBC, which accounts for 10–15% for BC, is the most lethal subtype with the highest recurrence rate in BC. Approximately 80% of TNBC are basal like breast cancer, which presents as invasive ductal carcinoma [2]. The poor prognosis of TNBC is potentially due to the lack of tumor-specific targeted therapy and the enrichment of cancer stem cell (CSC) populations, which are considered as driver of BC tumor progression, metastasis and therapeutic resistance [3]. CSCs have high self-renewal capacity and high plasticity, similar to pluripotent stem cells, with a consensus cell surface receptor signature, CD44CD24CD133ALDH1 [4]. Differentiation of CSCs is the source of cellular heterogeneity and tumor growth. Another feature of CSCs is their ablity to exit the cell cycle and enter a quiescence-like cell state known as dormancy [5]. Accumulating evidence indicate that breast cancer cell dormancy is the major cause of metastasis, therapeutic resistance and relapse. Dormant CSCs can remain undetected from immune surveillance and conventional therapies, such as radiotherapy and chemotherapy, leading to cancer recurrence even decades after initial diagnosis. The bone marrow (BM) is one of the most preferred sites of BC metastasis and cancer cell dormancy because of its unique microenvironment and instructive secretome from resident cells [6, 7]. Dormant CSCs in BM can form physical interactions with mesenchymal stem cells (MSCs) and hematopoietic supporting stromal cells, by forming gap junction-mediated intercellular communications (GJIC), which protect CSCs from the immune surveillance [8]. A better understanding of the molecular mechanisms underlying BC dormancy and recurrence remains as an essential requisite to improve clinical treatment.
In this review, we summarize some of the biological mechanisms underlying BC dormancy and recurrence. We also describe the interconnection between BC dormancy and tissue microenvironment, with particular focus on the BM niche.
Breast Cancer Dormancy
Tumor dormancy is a term created by Rupert A. Willis in 1934 who explained the delayed recurrence of cancer on metastatic sites after excision of the primary tumor. He stated that tumor cells must hide within the tissue in a dormant state, which can be awakened by environmental changes [9]. Decades after, this cancer dormant state was described as “temporary mitotic arrest” of tumor cells that could explain the long latency window (5 years) between the primary tumor removal and recurrence of the secondary tumor [10]. Based on this concept, minimal residual disease (MRD) can be considered as the source of dormant cancer cells [11]. MRD includes circulating tumor cells (CTCs) migrating through the circulatory or lymphatic systems, whereby a fraction of these cells known as disseminated tumor cells (DTCs) have the capacity of invading distant secondary sites, such as the BM in breast cancer [12]. Research including 10,307 BC patients shows that 27.3% of BC patients were DTC-positive in BM [13]. Moreover, accumulating evidence indicate that presence of DTCs in BM is highly correlated to a higher relapse rate and consequently higher death from breast cancer compared to DTC-negative BC patients [14–16].
Cancer dormancy can be specified into two models: (1) tumor mass dormancy, represented by constant and none growing small tumor masses, and (2) cellular dormancy, the quiescent or growth-arrested single and small DTC clusters [17]. Both models share similar characteristics including, mitotic arrest in a G-like state (quiescence) in response to external and internal signals, and their ability to maintain a balance between cell proliferation or cell death [11].
Tumor Microenvironment and Breast Cancer Metastasis
BCCs can undergo epithelial-mesenchymal transition (EMT) to generate CTCs, which can eventually colonize distant organs and develop into DTCs [18]. Hallmarks of EMT include reduction of epithelial cell markers such as E-cadherin (E-cad), -catenin, and zonula occludens-1 (ZO-1), along with the acquisition of mesenchymal markers such as N-cadherin, vimentin, and fibronectin [19]. Dormant BCCs can undergo EMT to generate CSCs, which may result in recurrence of BCCs populating their secondary microenvironment [20, 21]. The high plasticity of CSCs confers them the ability to switch between stem cell and non-stem cell states, along with other futures including self-renewal, clonal tumor initiation capacity, and long-term clonal repopulation potential [22]. CSCs can adapt to the microenvironment of tumor-distant tissues, such as BM, to form DTCs, where they may remain dormant for extended periods.
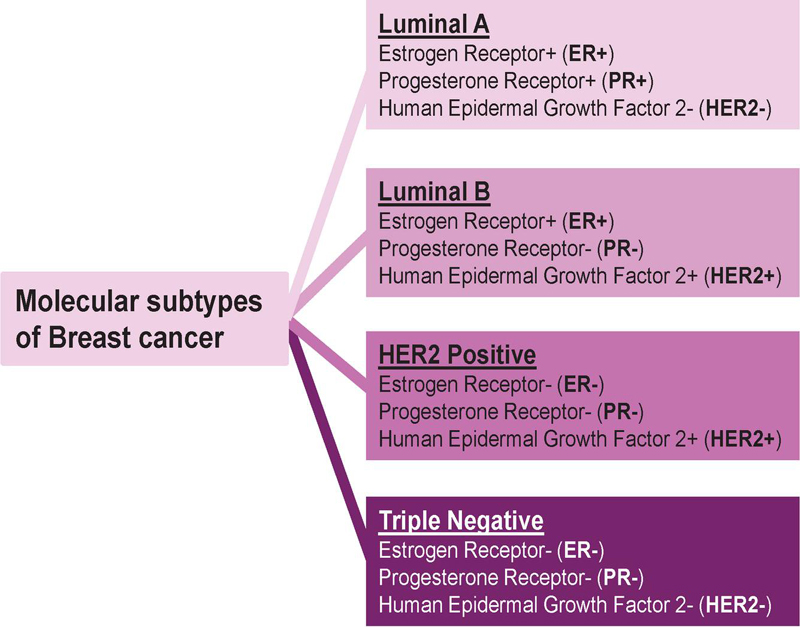
Figure 1 Molecular subtypes of breast cancer. Luminal A is positive for both estrogen (ER) and progesterone (PR) hormone receptors while negative for human epidermal growth factor 2 (HER2). Luminal B is ER, HER and PR. HER2 positive subtype is ER and PR. Triple negative subtype is ER, PR and HER.
Epithelial-mesenchymal plasticity (EMP) drive invasion, migration, and metastasis [23]. EMP determines ER BC dormancy, which is overcome by forced expression of E-cad [24]. EMP of BCCs can be regulated by the tumor microenvironment, which includes immune cells, stromal cells such as cancer-associated fibroblasts (CAFs), blood vessels, and extracellular matrix (ECM). CAFs are considered key mediators of BC metastasis [25]. These specialized fibroblasts surround the tumor mass modulate the ECM, secrete hormones, cytokines, and growth factors. For instance, CAFs-secreted C-X-C motif chemokine ligand (CXCL)-12, also known as stromal cell-derived factor-1 (SDF-1), promote BC progression and initiate EMT through the CXCL12-CXCR4 axis [26]. Additionally, CAF-secreted CXCL14 can induce nitric oxide synthase 1 (NOS1), leading to attenuated EMT and reduced migration of BCCs [27]. Furthermore, CAF-secreted transforming growth factor 1 (TGF-1) activates the TGF-/Smad signaling pathway in BCCs resulting in EMT initiation [28].
Another crucial regulator of EMT is tumor-associated macrophages (TAMs), which produce matrix proteolytic enzymes, including matrix metalloproteases (MMPs) such as MMP-9 and urokinase [29]. MMP-9 is a type of gelatinase that can degrade ECM, leading to the release of signaling molecules that promote invasion and migration of BCCs [30]. MMP-9 can activate the PI3K/Akt pathway in cancer cells, leading to the initiation of EMT [31]. Additionally, CXCL1 derived from TAMs promotes EMT and metastasis of BCCs via activation of the NF-B/SOX4 pathway [32].
Dormancy in Bone Marrow
Bone tissue is the most common site for breast cancer metastasis, which includes approximately 70% of metastatic BC patients [33]. This predilection for bone is due to the BM microenvironment, which supports BCC dormancy, as explained by Stephen Paget’s, “Seed and Soil” theory: ’When a plant goes to seed, its seeds are carried in all directions; but they can only live and grow if they fall on congenial soil’ [34]. There are two hematopoietic niches within the BM, the endosteal niche and the perivascular niche. The endosteal niche is characterized by bone-lining osteoblasts that maintain hematopoietic stem cell (HSC) quiescence, while the perivascular niche, located in the bone marrow stroma, is characterized by mesenchymal reticular and endothelial cells, which support mobilization and proliferation of HSCs [35].
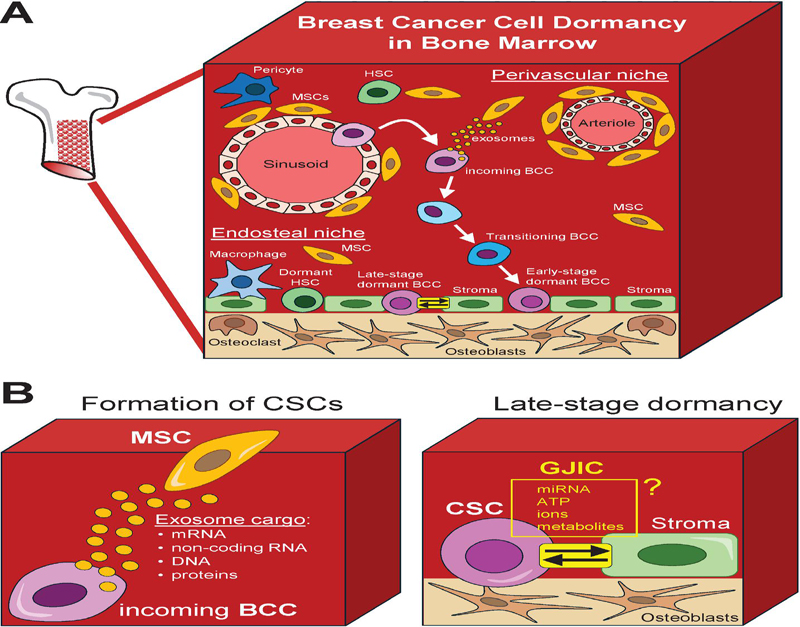
Figure 2 Breast cancer cell dormancy microenvironment in bone marrow. A) Schematic representation for an overview of the bone marrow (BM) microenvironment depicting the perivascular and endosteal niches supporting the generation of breast cancer stem cells (CSCs) and their transition into dormancy. Diverse interactions with bone marrow resident cells including mesenchymal stem cells (MSCs) support dedifferentiation of breast cancer cells (BCCs) into CSCs. B) Formation of CSCs. MSCs release exosome vesicles congaing a specific cargo (e.g., nucleic acids and proteins) to instructs incoming BCCs in the perivascular niche to dedifferentiate into CSCs. C) Late-stage dormancy. Direct interaction between CSCs and stroma cells via gap junction intercellular communication (GJIC) promotes CSC dormancy in the endosteum.
Dormant BCCs have been reported to reside within the stromal compartment, close to the endosteum [36]. BCC dormancy is facilitated by the establishment of connexin (Cx)-mediated GJIC between bone marrow stromal cells and BCCs, especially survived CSCs [37]. GJIC consists of clustered hemichannels that enabled direct intercellular communication, including the exchange of ions, organic molecules, microRNA (miRNA), and mitochondria between cells [38]. Cxs, which are a family of proteins with four transmembrane domains, are the molecular components of a hemichannel. GJIC is formed when two hemichannels from adjacent cells dock within an intercellular space of 2–3 nm [39]. CXCL12 has been known as a crucial regulator of GJIC formation. Upon establishing GJIC with BM stromal cells, these stromal cells pass CXCL12-specific miRNAs, including miR-127, -197, -222, and -223, to BCCs, leading to a reduction in CXCL12 production within BCCs [40]. This low concentration of CXCL12 can activate protein kinase C, which phosphorylates the gap junction protein Cx43, further enhancing GJIC [41]. Reduced CXCL12 levels prevents the formation of GJIC between BCCs and HSCs, thereby maintaining normal hematopoiesis [42].
MSCs, which are also hematopoietic supporting cells in the BM, can establish GJIC with BCCs [37]. Moreover, extracellular vesicles (EVs), including exosomes, secreted by MSCs, play a crucial role in regulating BCC dormancy. The presence of BCCs in the bone marrow can alter the content within the cargo of MSC-secreted exosomes. These EVs can shuttle cargo that includes microRNAs, mRNAs, proteins and DNA fragments into BCCs [43]. Such exosomal cargo can influence the physiology of BCCs. For instance, miR-23b in MSC-secreted exosomes induces a dormant phenotype in BCCs by suppressing MARCKS (myristoylated alanine rich protein kinase C substrate) gene encoding for a cytoplasmic membrane actin filament crosslinking protein that promotes cell cycling and motility [44]. During early stages of entry into the BM, BCCs can prime MSCs to release exosomes containing miR-222/223, which induce changes in the cell cycle promoting BCC quiescence [45]. Additionally, BCC-primed MSC-secreted EVs can trigger dedifferentiation of BCCs, particularly Oct4a BCCs, into CSCs, leading to early dormancy [46]. Beyond EVs, soluble signals from MSCs can also induce BCC dormancy. For example, NG2/Nestin MSCs produce TGF2 and BMP7, which activate quiescence in BCCs through TGFBRIII and BMPRII, leading to the activation of the SMAD, p38, and p27 pathways [47].
About 10% of BM stromal cells are M2 macrophages (Ms) and GJIC between M2 Ms and GJIC between M2 Ms and BCCs in leads to dormant phenotype of BCCs with cycling quiescence, reduced proliferation and carboplatin resistance. LPS-stimulated MSCs polarization of M2a Ms to M1 type. M1 M-derived exosomes activated NFêB to reverse quiescent BCCs to cycling cells, leading to the resurgence of breast cancer [48]. In BM, MSCs also provide immune protection to dormant BCCs by weakening CD8+ T cells and inducing immunosuppressive CD4IL-10 T cells and CD4CD25Foxp3 regulatory T cells [49–51].
Epigenetics in Breast Cancer Progression and Dormancy
Epigenetics is defined as heritable changes in gene expression without changing the DNA sequence [52]. Epigenetic dynamics are considered crucial mediators of CSC formation and potential cancer dormancy [53]. Epigenetic reprogramming, which leads to the activation of key cell survival gene networks, is required to initiate metastatic activation programs and dormancy. Epigenetic regulatory programs including DNA methylation, histone modifications, and non-coding RNAs (ncRNAs) controlling epigenetic factors are dysregulated in human BC and appear to show promising results as potential biomarkers for prognosis and for developing therapeutic treatments [43].
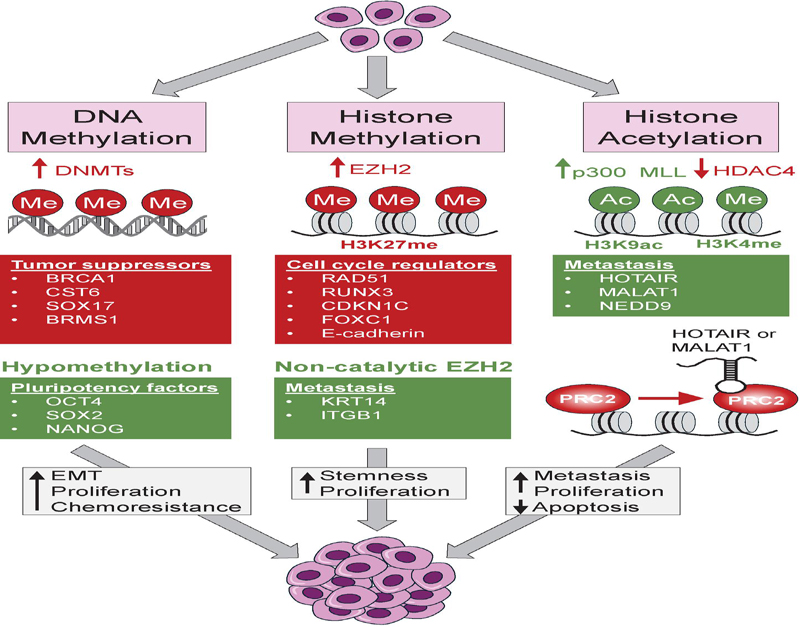
Figure 3 Epigenetic dysregulation in breast cancer. Upregulation of DNA methyltransferases (DNMTs) silence the expression of tumor suppressors including BRCA1, CST6, SOX17 and BRMS1. Conversely, DNA hypomethylation, also observed in BC, leads to the upregulation of pluripotency genes including OCT4, SOX2 and NANOG to promote epithelial-to-mesenchymal transitions (EMTs) and cellular stemness. Collectively, dysregulated DNA methylation leads to increased cellular proliferation and chemoresistance. Upregulation of the histone methyltransferase EZH2 cause an increased methylation of histone H3 at lysine 27 (H3K27me) resulting in downregulation of cell cycle regulators including RAD51, RUNX3, CDKN1C, FOXC1 and E-cadherin. A non-catalytic function of EZH2 promotes the expression of the metastatic promoting genes KRT14 and ITGB1. Dysregulated EZH2 levels and activity promote cellular stemness and proliferation. Upregulation of the histone acetyltransferase p300 and the histone methyltransferases MLL, along with downregulated HDAC4, is conducive to a transcriptionally permissive chromatin that favors the expression of the metastasis-promoting gene NEDD9 and the long-non-coding RNAs (lncRNAs) HOTAIR and MALAT1. These lncRNAs can reprogram the genomic recruitment of the polycomb repressive complex 2 (PRC2), which results in the upregulation of genes that stimulate EMT, invasiveness, metastasis and proliferation, along with downregulation of genes promoting apoptosis.
DNA methylation refers to the covalent attachment of methyl group(s) to the pyrimidine ring of the cytosine base within cytosine-phosphate-guanine (CpG) dinucleotides, generating 5-methylcytosine (5mC) [54]. The transfer of the methyl group(s) from the S-adenosyl-L-methionine (SAM) donor is catalyzed by DNA methyltransferases (DNMTs), including DNMT1, DNMT2, DNMT3A, and DNMT3B [55]. An active DNA demethylation process is catalyzed by the ten-eleven translocation (TET) family of enzymes TET1, TET2, TET3. These are dioxygenases [56]. While DNA methylation mediates the suppression of transcription, hypomethylation of DNA favors gene expression [57]. In BC, DNA hypermethylation was reported in key genes, such as the tumor suppressors BRCA1 and E-cadherin, the pro-apoptotic gene TMS1, and the transcription factor GATA3, whose expression correlates with BC patient survival [58–62]. The overexpression of DNMT3B is linked to hypermethylation of BRCA1, and the inhibition of DNMT activity by the dihydroxyflavanone compound liquiritigenin leads to increased BRCA1 transcriptional activity and decreased malignant behavior in TNBC [63, 64]. The expression levels of DNMTs are positively correlated with the methylation level of lactate dehydrogenase C4 (LDH-C4), which is associated with tumorigenesis and metastasis in diverse cancer types, including breast cancer [65]. DNA methylation works in concert with other epigenetic modifications. For example, DNMT1 inhibits the expression of the long non-coding RNA (lncRNA) PHACTR2-AS1 (PAS1), which mediates the silencing of PH20, an ECM modeler, via H3K9 methylation, to regulate cell migration. This DNMT1-PAS1-PH20 axis drives breast cancer growth and metastasis [66]. The interactions between DNMT1 and enhancer of zeste homolog-2 (EZH2), a histone methyltransferase that is part of the polycomb repressive complex 2 (PRC2), promote EMT in TNBC by silencing the gene WWC1, an active member of the Hippo/YAP pathway [67].
In BC, DNA methylation plays a crucial role in controlling the survival and cell fate of CTCs. Three important tumor suppressors – cystatin E/M (CST6), SRY-box transcription factor 17 (SOX17), and breast cancer metastasis suppressor gene 1 (BRMS1) – are hypermethylated in the CTCs of BC patients [68]. Furthermore, compared to single CTCs, the CTC clusters exhibit hypomethylated binding sites for stemness- and proliferation-associated transcription factors, including the pioneer pluripotency factors OCT4, NANOG, SOX2, and SIN3A, whose expression correlates with poor prognosis in BC patients [69]. Upregulation of OCT4 was shown in a most immature subset of BCCs that exhibit chemoresistance, stem cell properties, and dormancy [70]. The upregulation of NANOG triggers JAK/STAT and Wnt/b-catenin to induce stemness, self-renewal, metastasis, invasiveness and chemoresistance in diverse cancer types, including BC [71]. SOX2 is a CSC marker that stimulates EMT through Wnt/b-catenin leading to increased proliferation, invasion, metastasis, and drug resistance [3, 67]. Collectively, these finding suggest that formation and survival of CSCs via epigenetic programming is key for promoting BC metastasis and dormancy.
Additionally, histone modifications involved in BC development include methylation, acetylation, and phosphorylation of histone tails to induce the expression of specific genes that promote tumor growth and metastasis. For instance, the histone methyltransferase, enhancer of zeste homolog 2 (EZH2), catalyzes tri-methylation of lysine 27 on histone H3 (H3K27me3), which functions as a repressive epigenetic mark [72]. EZH2-mediated repression affects multiple cell cycle-related genes including RAD51, whose downregulation leads to activation of Raf1/pERK and b-catenin signaling pathways, RUNX3, resulting in low levels of the cell cycle arrest protein p21, CDKN1C, leading to accelerated G1-S cell cycle transition, FOXC1, a forkhead box transcription factor functioning as a negative regulator of cell migration and invasion, and CDH1 (E-cadherin), whose downregulation promotes metastasis [73]. Notably, in contrast to EZH2-H3K27me3 mediated gene repression, a non-catalytic form of EZH2 promotes the activation of the basal cytokeratin 14 (KRT14) gene, considered a hallmark in TNBC, by blocking its repressor SP1, which results in peritoneal metastasis [74]. The non-catalytic form of EZH2 also increases integrin subunit beta 1 (ITGB1), which subsequently activates TGF- signaling, promoting BC bone metastasis [75].
A global reduction in H4K16 acetylation levels is observed during the early stages of breast cancer [76]. In ER BCCs, ERs and ER-coregulators, including histone methyltransferases MLL1/MLL3 and histone acetyltransferase p300, induce the expression of HOX antisense intergenic RNA (HOTAIR), a lncRNA, which increases cell growth and viability of BC cells, thereby promoting breast cancer progression [77]. Additionally, histone deacetylases (HDACs) play a significant role in cancer by modifying the epigenome and consequently the expression of key breast cancer genes. For instance, the inhibition of HDAC4 leads to an upregulation of the NEDD9 gene, which induces cell proliferation and migration, thereby enhancing BC metastasis [78]. Downregulated HDAC11 in BC lymph node metastasis causes the upregulation of genes involved in promoting cancer cell migration to distal organs [79].
ncRNAs can also function as mediators of epigenetic dynamics in multiple cancer types [80]. ncRNAs have emerged as promising biomarkers for effectively distinguish diverse breast cancer subtypes, predict overall survival, and determine response to treatments [81]. ncRNAs are untranslated RNA molecules including miRNAs (miRs) and lncRNAs that can influence the expression of their target genes. For instance, miR-21 plays a crucial role in promoting invasive and metastatic BC by stimulating EMT and HIF1 expression in breast CSC-like cells [82]. The inhibition of miR-21 reverses such CSC signature and increases the susceptibility of BCCs to chemotherapy [83].
lncRNAs such as HOTAIR (HOX transcript antisense RNA) and MALAT1 (metastasis-associated lung adenocarcinoma transcript 1) contribute to tumor proliferation and metastatic progression in BC [84, 85]. In BC, HOTAIR reprograms the genomic occupancy of PRC2 towards an embryonic-like signature leading to the upregulation of genes involved in invasiveness and metastasis, while MALAT1 promotes the expression of genes involved in cell cycle progression and EMT. Additionally, upregulation of lncRNA NR2F1-AS1 in BCCs leads to the expression of pluripotency markers such as OCT4 and NANOG, as well as dormancy promoting genes inducing a quiescence-like state in ER BCCs [86]. Furthermore, the expression of ESR1 locus-enhancing and activating non-coding RNAs (ELEANORs) has been identified as a predictor of breast cancer recurrence in ER+ patients [87]. Mechanistically, ELEANORs upregulate the BC stem cell gene marker CD44, to maintain cellular stemness and possibly tumor dormancy.
Collectively, a comprehensive understanding of the epigenetic programs underlying BC progression, metastasis and relapse is critical for developing innovative treatments that can be focused on targeting dormant BCCs.
Glycolysis and OXPHOS Metabolism and Breast Cancer Dormancy
Cancer progression relies on the ability of cancer cells to rapidly fulfill their demand of amino acids, nucleotides and lipids, maintain redox homeostasis, and fuel their energy requirements [88]. This is one of the main reasons cancer cells are thought to upregulate the glycolytic pathway during the Warburg effect.
“Aerobic glycolysis” (or “Warburg glycolysis”) is characterized by increased glucose uptake and augmented lactate production even under conditions of normal oxygen levels [89]. In normal conditions, under constant oxygen supply, cells undergo glycolysis to produce pyruvate from glucose, then oxidized into carbon dioxide through oxidative phosphorylation (OXPHOS) in mitochondria, which produces a highly efficient energy yield [90]. However, under the condition of oxygen deprivation, healthy cells suffer incomplete oxidation of glucose resulting in lactate production, avoiding mitochondrial respiration, and consequently producing lower energy yield [91]. Cancer cells can reprogram their glucose metabolism by converting glucose into lactate via glycolysis, even in the presence of oxygen, though this mechanism is a much less efficient energy source [92]. Thus, cancer cells require alternative metabolic end products to accelerate their proliferation. Nevertheless, the role of aerobic glycolysis and OXPHOS in dormant BCCs remains undetermined.
Recent evidence supports the notion of metabolic reprogramming as another hallmark of cancer progression to keep up with energy demands during rapid proliferation. Part of this metabolic reprogramming includes lipid metabolism, which is increased in metastasis, as a source of energy even under hypoxic conditions. Lipid metabolism through fatty acid synthesis and glycerol is associated to BC dormancy [93]. In 2019, Schömel et al., studied glycolytic and oxidative metabolic phenotypes of BCCs following the overexpression of the glycosphingolipid UDP-glucose ceramide glycosyltransferase (UGCG), which is a key enzyme of lipid metabolism. UGCG activity results in the production of glycosylceramide, the precursor for all glycosphinglipids, and is related to poor prognosis in breast cancer patients [94]. This group showed that the overexpression of UGCG leads to increased proliferation of BCCs, which led them to investigate the molecular mechanism causing this proliferative advantage. Their following study demonstrated BCCs can fuel the tricarboxylic acid (TCA) cycle to sustain the proliferative capacity using glutathione production upon UGCG overexpression. Under this condition, BCCs experience a metabolic shift from a quiescent/aerobic state to an energetic state by increasing glycolysis and OXPHOS [95]. These studies highlight a close connection between BCC’s metabolic activity, glycosphingolipid metabolism and accordingly their dormant/active state. Although a strong focus on glycosphingolipid metabolism was presented, they also showed a higher BCC dependence on glycolysis during dormancy and attributed the BCC shift from a dormant to a proliferative state to be a consequence of increased OXPHOS. Therefore, this highlights the potential for dormant BCCs leveraging a reversed Warburg effect, using glycolysis to remain quiescent.
OXPHOS is considered as being the most efficient energy yielding metabolic pathway, with higher efficiency of adenosine triphosphate (ATP) production relative to glycolysis [96]. However, 60% of cancer cells are believed to rely on aerobic glycolysis for their energy supply [88]. While several studies have mainly focused on the impact of glycolysis on BCC dormancy, research also shows OXPHOS has energy source during BCC dormancy. It was recently shown that the microenvironment can contribute to a metabolic shift reliant on OXPHOS in the BM stroma. [97]. By using both the in vitro and in vivo models, the BCCs in the bone marrow exhibited an OXPHOS profile, along with low extracellular signal-regulated kinase (ERK) activation and high Akt signaling, two of the most activated signaling pathways in cancer by driving many cellular processes including cell proliferation, survival, and drug resistance [98].
A combination of both glycolysis and OXPHOS were suggested for BCC dormancy in a study by Sánchez-alvarez et al in 2020 [99]. This study shows the induction of BCC quiescence by overexpressing the mitochondrial fission factor (MFF) in MCF7s (ER+). Mitochondrial fission and fusion regulate the shape, function, and distribution of mitochondria, which is essential for maintaining cellular health, especially in response to metabolic demands, stress, and damage. MFF overexpression resulted in abnormal activated mitochondrial fission disrupting both glycolysis and OXPHOS and consequently stimulating quiescent programs. This result suggests mitochondrial fission could represent a tool in the activation of BCC dormancy.
Further studies are required to find whether glycolysis or OXPHOS is the main driver of BCC dormancy. In general, these studies have focused on the use of MCF7s (ER+) as a BC model. Since BC is a multifaceted disease, an exploration of pathways feeding these two key metabolic processes within different BC subtypes is needed. In summary, glycolysis and OXPHOS play crucial roles in the maintenance of BC dormancy. The ability of cancer cells to switch between these two pathways confers them with the cellular plasticity for adapting to diverse tumor microenvironments and contribute to their long-term survival and potential for relapse. Therefore, targeting these metabolic processes could provide new avenues for new therapeutic developments to prevent BC recurrence.
Hypoxia and Breast Cancer Dormancy
Low oxygen levels influence cellular metabolism, which affects proliferation, migration, and invasion. Cells are evolutionarily programmed to engage in glycolysis when oxygen levels are too low to support OXPHOS [100]. Hypoxia is a hallmark of tumor microenvironment in most solid tumor, which is caused by an imbalance between oxygen consumption and supply due to rapid tumor growth [101]. Hypoxia promotes tumor growth, metastasis, and drug resistance by regulating the expression of hypoxia-related genes, eventually leading to more aggressive cancers [102]. The hypoxia-inducible factor (HIF) family of transcriptional factors play a pivotal role in the cellular response to hypoxic stress. HIF1 (hypoxia-inducible factor 1 alpha), which affects the expression of specific gene networks, is involved in cellular energetics and metabolism [103, 104]. Part of the tumor microenvironment is composed of hypoxic regions which favors cancer cells to undergo cell cycle quiescence and chemotherapeutic resistance [90]. However, in a dormant cancer model, that is not within a solid tumor, dormant BCCs exhibit cycling quiescence and chemoresistance by existing as single cells at metastatic sites such as BM [105].
In hypoxic environment, oxygen deprivation can alter epigenetic regulatory programs in cancer cells causing increased metastatic competence and facilitating cellular transitions towards stem cell-like states [106]. Epigenetic mechanisms in hypoxic responses during cancer dormancy in BM was described by Ferrer et al [90]. Some of these epigenetic reprogramming mechanisms include DNA methylation-demethylation dynamics facilitating cell fate changes towards CSC-like states. Hypoxia-driven epigenetic changes can promote the formation of CSCs [36]. Under hypoxic conditions, hypermethylation of promoter regions and downregulation of TET activity can silence the expression of tumor suppressor genes and contribute to BC dormancy [107].
In summary, hypoxia may facilitate BC dormancy by inducing a quiescent state while maintaining CSCs. Targeting the hypoxic microenvironment and the related signaling pathways could provide new therapeutic chances to prevent cancer recurrence and improve patient outcomes.
Wnt Signaling and Breast Cancer Dormancy
Wnt signaling regulates a variety of cellular processes such as cell fate, differentiation, proliferation, and stem cell pluripotency. Aberrant Wnt signaling is a hallmark of multiple cancer types, particularly an aggressive subtype of BC, known as triple-negative breast cancer (TNBC) [108]. Wnt signaling plays a critical role in BC dormancy by promoting cell survival in a quiescent state and potentially contribute to late recurrence. Using HER2 transgenic mice as BC model, studies showed that BCCs can disseminate in tumor premalignant stage, and EMT induced by both canonical and noncanonical Wnt signaling contributes to early dissemination [109]. These early disseminated cells show stem-cell like characteristics and have tumor-initiating capacity. Although quiescent, they can release themselves from dormancy, become migratory and invasive, leading to distal metastasis. They are defined as metastatic-initiating cells (MIC) and this may be responsible for tumor metastasis even years after removal of primary tumors [110]. Notably, one recent study showed that during dormancy, DKK1 (Dickkopf Wnt signaling pathway inhibitor (1) suppresses Wnt signaling in MIC. This was identified as latent competent cancer (LCC) and downregulation of Wnt signaling induces LCC to quiescence to evade immune surveillance [111]. Therefore, these findings propose that after early MIC dissemination, Wnt signaling activity may decline for subsequent MIC dormancy. Understanding how Wnt signaling interacts with other cellular processes during BC dormancy is key for developing strategies to prevent relapse in BC patients.
Therapeutic Treatments
Breast cancer dormancy is a major clinical problem. Most current therapies are unable to target non-dividing dormant cancer cells. Thus, one therapeutic approach is to force dormant cancer cells to exit their quiescent stage followed by conventional therapies targeting highly proliferating cells. However, this approach depends on how much of the proliferating cells can be removed as they escape dormancy. Failure to this approach could increase the chance of metastatic latency [112]. Although effective strategies to target dormant cancer cells have been developed, potential issues may occur. For instance, DTC assessment requires painful biopsies, and even after successful evaluation, methods to eliminate of DTCs remain to be developed.
Clinical trials can target both dormant and active BCCs. ER+ BC trials that targets the upregulation of the cyclin-dependent kinase 4/5 pathway, which have been shown to influence BCC dormancy exit have been described. The goal for this clinical trial is to target metabolic pathways in both dormant BCCs and active BCCs (clinical trial identifier: NCT04841148). In addition, mitochondrial ATP-depletion therapy may be used to prevent BCC dormancy [113]. Additionally, it is critical to focus on cancer cell growth arrest [114]. For instance, the cell proliferation marker Ki67, which is not expressed during G0 phase of cell cycle, could be used as BC marker [115].
Collectively, addressing BC dormancy involves understanding the mechanisms that allow cancer cells to remain viable in a quiescent state to develop new therapies specifically targeting dormant cells. Due to the multifactorial programs supporting BC dormancy, including microenvironmental conditions, epigenetics, metabolism, and cell signaling, it seems logical that new therapies will have to depend on the combination of multiple targeting approaches to improve BC patient outcomes.
References
[1] Barzaman, K., et al., Breast cancer: Biology, biomarkers, and treatments. Int Immunopharmacol, 2020. 84: p. 106535.
[2] Zagami, P. and L.A. Carey, Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer, 2022. 8(1): p. 95.
[3] Zhang, X., K. Powell, and L. Li, Breast Cancer Stem Cells: Biomarkers, Identification and Isolation Methods, Regulating Mechanisms, Cellular Origin, and Beyond. Cancers (Basel), 2020. 12(12).
[4] Talukdar, S.B., P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B., Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res., 2019. 141: p. 43–84.
[5] Ferrer, A.I., Trinidad, J.R., Sandiford, O. et al., Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev, 2020. 39: p. 721–738.
[6] Sowder, M.E., and Rachelle W Johnson, Bone as a Preferential Site for Metastasis. JBMR, 2019. 3(3).
[7] Eltoukhy, H.S., et al., Secretome within the bone marrow microenvironment: A basis for mesenchymal stem cell treatment and role in cancer dormancy. Biochimie, 2018. 155: p. 92–103.
[8] Walker, N.D., et al., The bone marrow niche in support of breast cancer dormancy. Cancer Lett, 2016. 380(1): p. 263–71.
[9] Willis, R.A., The Spread of Tumours in the Human Body. 1934, London: J. and A. Churchill.
[10] Hadfield, G., The dormant cancer cell. Br Med J, 1954. 2(4888): p. 607–10.
[11] Dalla, E., et al., Dormancy in Breast Cancer. Cold Spring Harb Perspect Med, 2023. 13(11).
[12] Tachtsidis, A., et al., Minimal residual disease in breast cancer: an overview of circulating and disseminated tumour cells. Clin Exp Metastasis, 2016. 33(6): p. 521–50.
[13] Hartkopf, A.D., et al., Disseminated tumour cells from the bone marrow of early breast cancer patients: Results from an international pooled analysis. Eur J Cancer, 2021. 154: p. 128–137.
[14] Bidard, F.C., et al., Disseminated tumor cells of breast cancer patients: a strong prognostic factor for distant and local relapse. Clin Cancer Res, 2008. 14(11): p. 3306–11.
[15] Hartkopf, A.D., et al., Disseminated tumor cells from the bone marrow of patients with nonmetastatic primary breast cancer are predictive of locoregional relapse. Ann Oncol, 2015. 26(6): p. 1155–1160.
[16] Wiedswang, G., et al., Detection of isolated tumor cells in bone marrow is an independent prognostic factor in breast cancer. J Clin Oncol, 2003. 21(18): p. 3469–78.
[17] Aguirre-Ghiso, J.A., Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer, 2007. 7(11): p. 834–46.
[18] Weidenfeld, K. and D. Barkan, EMT and Stemness in Tumor Dormancy and Outgrowth: Are They Intertwined Processes? Front Oncol, 2018. 8: p. 381.
[19] Reddy, B.Y., et al., The Microenvironmental Effect in the Progression, Metastasis, and Dormancy of Breast Cancer: A Model System within Bone Marrow. Int J Breast Cancer, 2012. 2012: p. 721659.
[20] Casson, J., et al., Mesenchymal stem cell-derived extracellular vesicles may promote breast cancer cell dormancy. J Tissue Eng, 2018. 9: p. 2041731418810093.
[21] Mani, S.A., et al., The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 2008. 133(4): p. 704–15.
[22] Plaks, V., N. Kong, and Z. Werb, The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell, 2015. 16(3): p. 225–38.
[23] Brabletz, S., et al., Dynamic EMT: a multi-tool for tumor progression. EMBO J, 2021. 40(18): p. e108647.
[24] Aouad, P., et al., Epithelial-mesenchymal plasticity determines estrogen receptor positive breast cancer dormancy and epithelial reconversion drives recurrence. Nat Commun, 2022. 13(1): p. 4975.
[25] Li, Y., et al., The role of cancer-associated fibroblasts in breast cancer metastasis. Front Oncol, 2023. 13: p. 1194835.
[26] Butti, R., et al., Tumor-derived osteopontin drives the resident fibroblast to myofibroblast differentiation through Twist1 to promote breast cancer progression. Oncogene, 2021. 40(11): p. 2002–2017.
[27] Sjoberg, E., et al., A Novel ACKR2-Dependent Role of Fibroblast-Derived CXCL14 in Epithelial-to-Mesenchymal Transition and Metastasis of Breast Cancer. Clin Cancer Res, 2019. 25(12): p. 3702–3717.
[28] Yu, Y., et al., Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br J Cancer, 2014. 110(3): p. 724–32.
[29] Mehta, A.K., et al., Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front Immunol, 2021. 12: p. 643771.
[30] Jacob, A. and R. Prekeris, The regulation of MMP targeting to invadopodia during cancer metastasis. Front Cell Dev Biol, 2015. 3: p. 4.
[31] Tian, K., et al., MMP-9 secreted by M2-type macrophages promotes Wilms’ tumour metastasis through the PI3K/AKT pathway. Mol Biol Rep, 2022. 49(5): p. 3469–3480.
[32] Wang, N., et al., CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis, 2018. 9(9): p. 880.
[33] Pulido, C., et al., Bone metastasis risk factors in breast cancer. Ecancermedicalscience, 2017. 11: p. 715.
[34] Paget, S., The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev, 1989. 8(2): p. 98–101.
[35] Ema, H. and T. Suda, Two anatomically distinct niches regulate stem cell activity. Blood, 2012. 120(11): p. 2174–81.
[36] Patel, S.A., et al., Delineation of breast cancer cell hierarchy identifies the subset responsible for dormancy. Sci Rep, 2012. 2: p. 906.
[37] Sinha, G., et al., Gap Junctions and Breast Cancer Dormancy. Trends Cancer, 2020. 6(4): p. 348–357.
[38] Aasen, T., et al., Gap junctions and cancer: communicating for 50 years. Nat Rev Cancer, 2016. 16(12): p. 775–788.
[39] Oshima, A., Structure and closure of connexin gap junction channels. FEBS Lett, 2014. 588(8): p. 1230–7.
[40] Lim, P.K., et al., Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res, 2011. 71(5): p. 1550–60.
[41] Park, J.M., et al., Exogenous CXCL12 activates protein kinase C to phosphorylate connexin 43 for gap junctional intercellular communication among confluent breast cancer cells. Cancer Lett, 2013. 331(1): p. 84–91.
[42] Moharita, A.L., et al., SDF-1alpha regulation in breast cancer cells contacting bone marrow stroma is critical for normal hematopoiesis. Blood, 2006. 108(10): p. 3245–52.
[43] Ferrer, A.I., et al., Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev, 2020. 39(3): p. 721–738.
[44] Ono, M., et al., Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signal, 2014. 7(332): p. ra63.
[45] Bliss, S.A., et al., Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res, 2016. 76(19): p. 5832–5844.
[46] Sandiford, O.A., et al., Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res, 2021. 81(6): p. 1567–1582.
[47] Nobre, A.R., et al., Bone marrow NG2(+)/Nestin(+) mesenchymal stem cells drive DTC dormancy via TGFbeta2. Nat Cancer, 2021. 2(3): p. 327–339.
[48] Walker, N.D., et al., Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death Dis, 2019. 10(2): p. 59.
[49] Montesinos, J.J., et al., In vitro evidence of the presence of mesenchymal stromal cells in cervical cancer and their role in protecting cancer cells from cytotoxic T cell activity. Stem Cells Dev, 2013. 22(18): p. 2508–19.
[50] Hsu, W.T., et al., Prostaglandin E2 potentiates mesenchymal stem cell-induced IL-10+IFN-gamma+CD4+ regulatory T cells to control transplant arteriosclerosis. J Immunol, 2013. 190(5): p. 2372–80.
[51] Bernardo, M.E. and W.E. Fibbe, Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell, 2013. 13(4): p. 392–402.
[52] Waddington, C.H., The epigenotype. 1942. Int J Epidemiol, 2012. 41(1): p. 10–3.
[53] Crea, F., et al., The epigenetic/noncoding origin of tumor dormancy. Trends Mol Med, 2015. 21(4): p. 206–11.
[54] Moore, L.D., T. Le, and G. Fan, DNA methylation and its basic function. Neuropsychopharmacology, 2013. 38(1): p. 23–38.
[55] Lyko, F., The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet, 2018. 19(2): p. 81–92.
[56] Lio, C.J., et al., TET methylcytosine oxidases: new insights from a decade of research. J Biosci, 2020. 45.
[57] Suva, M.L., N. Riggi, and B.E. Bernstein, Epigenetic reprogramming in cancer. Science, 2013. 339(6127): p. 1567–70.
[58] Hurtado, A., et al., FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet, 2011. 43(1): p. 27–33.
[59] Mirza, S., et al., Promoter hypermethylation of TMS1, BRCA1, ERalpha and PRB in serum and tumor DNA of invasive ductal breast carcinoma patients. Life Sci, 2007. 81(4): p. 280–7.
[60] Shargh, S.A., et al., Downregulation of E-cadherin expression in breast cancer by promoter hypermethylation and its relation with progression and prognosis of tumor. Med Oncol, 2014. 31(11): p. 250.
[61] Theodorou, V., et al., GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res, 2013. 23(1): p. 12–22.
[62] Zhu, X., et al., Hypermethylation of BRCA1 gene: implication for prognostic biomarker and therapeutic target in sporadic primary triple-negative breast cancer. Breast Cancer Res Treat, 2015. 150(3): p. 479–86.
[63] Esteller, M., et al., Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst, 2000. 92(7): p. 564–9.
[64] Liang, F., et al., Liquiritigenin decreases tumorigenesis by inhibiting DNMT activity and increasing BRCA1 transcriptional activity in triple-negative breast cancer. Exp Biol Med (Maywood), 2021. 246(4): p. 459–466.
[65] Zhang, J., et al., Correlation between promoter methylation of the LDH-C4 gene and DNMT expression in breast cancer and their prognostic significance. Oncol Lett, 2022. 23(1): p. 35.
[66] Fu, Y., et al., The DNMT1-PAS1-PH20 axis drives breast cancer growth and metastasis. Signal Transduct Target Ther, 2022. 7(1): p. 81.
[67] Liu, X., et al., The EZH2- H3K27me3-DNMT1 complex orchestrates epigenetic silencing of the wwc1 gene, a Hippo/YAP pathway upstream effector, in breast cancer epithelial cells. Cell Signal, 2018. 51: p. 243–256.
[68] Chimonidou, M., et al., DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin Chem, 2011. 57(8): p. 1169–77.
[69] Gkountela, S., et al., Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell, 2019. 176(1–2): p. 98–112 e14.
[70] Shyam A. Patel, S.H.R., Margarette Bryan, Lillian F. Pliner, Gabriela Dontu, Prem S. Patel, Sohrab Amiri, Sharon R. Pine, and Pranela Rameshwar, Delineation of breast cancer cell hierachy identifies the subset responsible for dormancy. 2012.
[71] Vasefifar, P., et al., Nanog, as a key cancer stem cell marker in tumor progression. Gene, 2022. 827: p. 146448.
[72] Kim, K.H. and C.W. Roberts, Targeting EZH2 in cancer. Nat Med, 2016. 22(2): p. 128–34.
[73] Yoo, K.H. and L. Hennighausen, EZH2 methyltransferase and H3K27 methylation in breast cancer. Int J Biol Sci, 2012. 8(1): p. 59–65.
[74] Verma, A., et al., EZH2-H3K27me3 mediated KRT14 upregulation promotes TNBC peritoneal metastasis. Nat Commun, 2022. 13(1): p. 7344.
[75] Zhang, L., et al., EZH2 engages TGFbeta signaling to promote breast cancer bone metastasis via integrin beta1-FAK activation. Nat Commun, 2022. 13(1): p. 2543.
[76] Guo, P., et al., The Histone Acetylation Modifications of Breast Cancer and their Therapeutic Implications. Pathol Oncol Res, 2018. 24(4): p. 807–813.
[77] Bhan, A., et al., Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol Biol, 2013. 425(19): p. 3707–22.
[78] Hu, Z., et al., Histone deacetylase inhibitors promote breast cancer metastasis by elevating NEDD9 expression. Signal Transduct Target Ther, 2023. 8(1): p. 11.
[79] Leslie, P.L., et al., Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat Commun, 2019. 10(1): p. 4192.
[80] Kumar, S., et al., Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers (Basel), 2020. 12(12).
[81] Panjarian, S. and J.J. Issa, The Roles of DNA Demethylases in Triple-Negative Breast Cancer. Pharmaceuticals (Basel), 2021. 14(7).
[82] Han, M., et al., MiR-21 regulates epithelial-mesenchymal transition phenotype and hypoxia-inducible factor-1alpha expression in third-sphere forming breast cancer stem cell-like cells. Cancer Sci, 2012. 103(6): p. 1058–64.
[83] Mei, M., et al., Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol Cancer Res Treat, 2010. 9(1): p. 77–86.
[84] Gupta, R.A., et al., Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature, 2010. 464(7291): p. 1071–6.
[85] Jadaliha, M., et al., Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget, 2016. 7(26): p. 40418–40436.
[86] Sanchez Calle, A., et al., Long non-coding NR2F1-AS1 is associated with tumor recurrence in estrogen receptor-positive breast cancers. Mol Oncol, 2020. 14(9): p. 2271–2287.
[87] Fukuoka, M., et al., The ELEANOR noncoding RNA expression contributes to cancer dormancy and predicts late recurrence of estrogen receptor-positive breast cancer. Cancer Sci, 2022. 113(7): p. 2336–2351.
[88] Hanahan, D. and R.A. Weinberg, The hallmarks of cancer. Cell, 2000. 100(1): p. 57–70.
[89] Pranzini, E., G. Raugei, and M.L. Taddei, Metabolic Features of Tumor Dormancy: Possible Therapeutic Strategies. Cancers (Basel), 2022. 14(3).
[90] Ferrer, A., et al., Hypoxia-mediated changes in bone marrow microenvironment in breast cancer dormancy. Cancer Lett, 2020. 488: p. 9–17.
[91] Martins Pinto, M., et al., The Warburg effect and mitochondrial oxidative phosphorylation: Friends or foes? Biochim Biophys Acta Bioenerg, 2023. 1864(1): p. 148931.
[92] Koppenol, W.H., P.L. Bounds, and C.V. Dang, Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer, 2011. 11(5): p. 325–37.
[93] Ruidas, B., Mitochondrial lipid metabolism in metastatic breast cancer. 2024.
[94] Schomel, N., et al., UGCG influences glutamine metabolism of breast cancer cells. Sci Rep, 2019. 9(1): p. 15665.
[95] Schomel, N., et al., UGCG overexpression leads to increased glycolysis and increased oxidative phosphorylation of breast cancer cells. Sci Rep, 2020. 10(1): p. 8182.
[96] Zheng, J., Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett, 2012. 4(6): p. 1151–1157.
[97] Buschhaus, J.M., et al., Targeting disseminated estrogen-receptor-positive breast cancer cells in bone marrow. Oncogene, 2020. 39(34): p. 5649–5662.
[98] Martini, M., et al., PI3K/AKT signaling pathway and cancer: an updated review. Ann Med, 2014. 46(6): p. 372–83.
[99] Sanchez-Alvarez, R., et al., Mitochondrial Fission Factor (MFF) Inhibits Mitochondrial Metabolism and Reduces Breast Cancer Stem Cell (CSC) Activity. Front Oncol, 2020. 10: p. 1776.
[100] Tau, S. and T.W. Miller, The role of cancer cell bioenergetics in dormancy and drug resistance. Cancer Metastasis Rev, 2023. 42(1): p. 87–98.
[101] Munoz, D., et al., Effects of screening and systemic adjuvant therapy on ER-specific US breast cancer mortality. J Natl Cancer Inst, 2014. 106(11).
[102] Wang, W., et al., Hypoxia-inducible factor 1alpha in breast cancer prognosis. Clin Chim Acta, 2014. 428: p. 32–7.
[103] Paredes, F., H.C. Williams, and A. San Martin, Metabolic adaptation in hypoxia and cancer. Cancer Lett, 2021. 502: p. 133–142.
[104] Alejandra Ferrer, J.R.T., Oleta Sandiford,, Jean-Pierre Etchegaray, and Pranela Rameshwar, Epigenetic dynamics in cancer stem cell dormancy. 2020.
[105] Davis, J.E., Jr., et al., Tumor Dormancy and Slow-Cycling Cancer Cells. Adv Exp Med Biol, 2019. 1164: p. 199–206.
[106] Chen, A., et al., Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene, 2018. 37(31): p. 4214–4225.
[107] Thienpont, B., et al., Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature, 2016. 537(7618): p. 63–68.
[108] Pohl, S.G., et al., Wnt signaling in triple-negative breast cancer. Oncogenesis, 2017. 6(4): p. e310.
[109] Harper, K.L., et al., Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature, 2016. 540(7634): p. 588–592.
[110] Yin, P., et al., Wnt signaling in human and mouse breast cancer: Focusing on Wnt ligands, receptors and antagonists. Cancer Sci, 2018. 109(11): p. 3368–3375.
[111] Malladi, S., et al., Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell, 2016. 165(1): p. 45–60.
[112] Bushnell, G.G., et al., Breast cancer dormancy: need for clinically relevant models to address current gaps in knowledge. NPJ Breast Cancer, 2021. 7(1): p. 66.
[113] Fiorillo, M., et al., High ATP Production Fuels Cancer Drug Resistance and Metastasis: Implications for Mitochondrial ATP Depletion Therapy. Front Oncol, 2021. 11: p. 740720.
[114] Nielsen, T.O., et al., Assessment of Ki67 in Breast Cancer: Updated Recommendations From the International Ki67 in Breast Cancer Working Group. J Natl Cancer Inst, 2021. 113(7): p. 808–819.
[115] Bartlome, S. and C.C. Berry, Recent insights into the effects of metabolism on breast cancer cell dormancy. Br J Cancer, 2022. 127(8): p. 1385–1393.
International Journal of Translational Science, Vol. 1, 303–326.
doi: 10.13052/ijts2246-8765.2024.042
© 2025 River Publishers