An Overview of the Biological Processes of Mesenchymal Stem Cells and Their Response to an Inflammatory Milieu
Lauren S. Sherman, Marianne D. Castillo, Monique H. Johnson and Pranela Rameshwar*
Rutgers New Jersey Medical School, Department of Medicine, Newark, NJ, USA
E-mail: shermala@njms.rutgers.edu; rameshwa@njms.rutgers.edu
*Corresponding Author
Received 25 April 2025; Accepted 02 May 2025
Abstract
There is continued interest in applying adult human mesenchymal stem cells (MSCs) in tissue regeneration, as well as mitigating overt inflammation such as graft versus host disease. MSCs are derived from different sources such as bone marrow, bone chips, dental pulp, adipose tissues, and placenta. MSCs appear to exert a memory for their tissue of origin. Interestingly, despite positive outcomes of MSCs in vivo, their survival in host tissues appears to be short-lived. To date, based on research and clinical literature, there is a need to understand how MSCs are maintained in a multipotent state. We discuss the role for the inflammatory milieu in which soluble and insoluble such as exosomes can determine the immune response of MSCs – suppressor versus enhancer. This article discusses a central role for the transcription factor NFB in maintaining multipotency, and discusses how the inflammatory milieu influences the differentiation of MSCs. Lessons are drawn from the literature on MSCs in cancer to further describe the role of NFB in maintaining the stem cell state. The findings outlined in this article could be important to future translational studies.
Keywords: Stem cells, cytokines, inflammation, NFB, purinergic receptors, multipotency.
Mesenchymal Stem Cells (MSCs) – Brief Introduction
Stem cells hold great promise for treating a variety of pathological conditions, ranging from drug delivery, anti-inflammatory conditions, and regenerative medicine [1–4]. MSCs are multi-potent, non-hematopoietic adult stem cells with regenerative and unique immune properties. First discovered in the bone marrow by Alexander Friedenstein in the 1960s, MSCs have now been identified in multiple adult and fetal tissues. In adults, MSCs are found in tissues such as adipose, bone, bone marrow, dental pulp, and placenta [5–9].
Although derived from different tissues, MSCs share key characteristics including adherence to plastics, self-renewal, and multilineage differentiation (e.g. adipocytes, osteocytes, and chondrocytes). MSCs can home to areas of inflammation and can cross the blood–brain barrier. In an inflammatory milieu, MSCs are licensed or educated by the inflammatory mediators to be immune-suppressor cells. MSCs are generally characterized by specific cell surface proteins – CD34, CD45, HLA-DR, CD44, CD73, CD90, CD105, STRO-1 [2, 10, 11].
MSCs are functionally plastic due to their ability to generate cells of various tissues such as bone, fat, muscle, cartilage, myocytes and neurons [12, 13]. MSCs are also associated with multiple functions including angiogenesis, homing to areas of inflammation, and systemic modulation of immune responses – pro-anti-inflammatory. These inflammatory functions depend on the microenvironment that could be cytokines, neuropeptides and microvesicles [14–19].
MSCs can produce baseline and can be induced to produce cytokines. The types of cytokines produced by MSCs depend on the cues within the tissue niche [14–17]. MSCs can respond to the cytokines due to multiple cytokine and chemokine receptors. As third-party cells within an inflammatory milieu such as graft versus host disease (GvHD), MSCs can be licensed as immune suppressor cells to modulate the inflammation [14]. Minor differences in phenotype and function have been attributed to MSCs derived from different tissue sources, but the key functions of MSCs as listed above are shared by MSCs derived from all sources [20, 21]. Critically, MSCs can cross allogeneic barriers, permitting MSCs derived from one individual to another, hence the potential to be used as an off-the-shelf stem cell source [1, 14].
The source of MSCs for expansion or direct use can be accomplished through minimally invasive procedures including bone marrow aspiration, abdominoplasty, lipoaspiration, and collecting of other discard tissues such as the placenta [18, 22]. The isolated tissues can be used in tissue regeneration using although this will result in the use of heterogeneous cells. Most applications use enriched MSCs that are achieved in culture. Together, the ease of isolation, capacity to cross allogeneic barrier, evidence of safety, and reduced ethical concerns explain MSCs’ clinical appeal.
To date, MSCs are listed in over six thousand clinical trials (clinicaltrials.gov, July 1, 2023), capitalizing on the properties of MSCs described throughout this article – primarily their ability to home to areas of inflammation and to reduce inflammation locally and/or systemically [1, 3, 4, 14, 23–26]. Beyond these currently listed applications, MSCs are being studied as potential therapy in conditions such as cancer, diabetes mellitus, and end-stage liver disease [1–3, 26].
Homing of MSCs
Migration of MSCs from their microenvironmental niche or site of administration to the target tissue or organ is a fundamental characteristic of MSCs that facilitates their response to inflammation and tissue regeneration. MSC homing, which refers to MSCs migration towards a target, is facilitated by cytokines and chemokines released at the inflammatory site [18, 25]. To reiterate, MSCs express receptors that can interact with multiple cytokines and chemokines, which permits them to respond to an inflammatory microenvironment, including migration and response to rapidly changing microenvironments [27, 28]. Of note, MSCs express CXCR4 and CXCR7, which are receptors for CXCL12/SDF1, which permits their chemoattraction to sites of inflammation [29]. Other chemokines have also been identified to serve as chemoattraction of MSCs [30–32]. An understanding of how MSCs are attracted to sites of inflammation is well-studied since insights into the mechanisms would allow for targeted therapy. Such treatment could be relevant to the case of immune regulation, and as a mechanism to deliver drugs [33]. Once MSCs migrate to sites of inflammation, they can be directly involved in tissue regeneration. However, the role of MSCs in tissue regeneration has been predominantly due to their secretome [1, 34, 35]. The efficiency of exogenous MSC mobilization to the target tissue significantly impacts the efficacy of these cells to act as a possible therapy for diseases such as degenerative disorders and immune modulators [4].
Despite the promise of MSCs in medicine, their mobilization is inefficient (10%), leading to debate over optimal routes of administration to improve mobilization for different applications [35]. Local administration of MSCs to the target organ or tissue is one route that may enhance the efficacy of these stem cells. However, this mode of delivering MSC can be highly invasive, resulting in the probability of the MSCs escaping the [36]. Intravenous administration has also been suggested. However, most of the cells administered intravenously are entrapped in the lung due to the MSCs’ large size and their expression of adhesion molecules such as VCAM1 and VLA-4, which can bind lung epithelial which can bind lung epithelial cells. It is estimated that only 1% of the cells reach the target organ [37–39]. The half-life of these entrapped MSCs is short – under four days – supporting a role for MSC secretome in the clinical effects of MSCs [40–42].
Different molecular engineering approaches have been employed to increase MSC homing to specific sites, including overexpression of cytokine receptors and downregulation of adhesion molecules [43–48]. Another approach shown to enhance MSC homing is magnetic guidance to the target tissue, whereby the MSCs are labeled with magnetic carbon nanotubules prior to administration [49]. Notably these studies have largely found decreased lung entrapment and increased homing of MSCs to the target tissues without significant effect on the viability of the cells or their capacity to differentiate [45, 49]. It is crucial to keep in mind that enhanced homing of MSCs and their response requires different approaches with the targeted organ or tissue as guide to the approach. Furthermore, the specific disease being treated, and the source and pre-treatment of the MSCs are also factors when designing treatment. Thus, understanding these nuances is key to developing effective treatments and improving patient outcomes.
MSCs – Source of Endocrine and Paracrine Mediators in Intercellular Communication
As discussed above, there is little evidence to support the basis of the clinical responses of MSCs, with respect to tissue engraftment. This statement is based on a small percentage of MSCs that reaches the target organ with no strong evidence of long-term survival a mechanism by which MSCs facilitate tissue repair is likely due to released soluble factors, to released soluble factors, including cytokines and extracellular vesicles (EVs). Also, upon injection or engraftment. stem cells are deprived of their extracellular environment. This deprived the stem cells of soluble as well as physical, chemical, and biochemical factors that are needed for engraftment. In this context, soluble factors play a fundamental, but not exclusive to engraftment.
Among classes of EVs, exosomes are the most investigated. Exosomes are MVs of 30–150 nm in diameter, enriched in proteins, miRNA, lipids, and other regulatory molecules. They have been identified in a wide array of body fluids such as blood (including plasma and serum), urine, saliva, cerebrospinal fluid, pleural effusion, and ascites, and are considered representative of the cells from which they were derived (discussed in [50]). Due to their source-cell representation, circulating MVs are under investigation for diagnostic purposes, evaluating biomarkers for the presence and type of aberrant cells – including malignancy, injury, and other disorders [50–53]. It thus stands to reason that the MVs derived from MSCs of different source tissues, and different treatments and exposures in vitro, would differ from one another [54, 55]. Due to the similarity of MSCs and their derived exosomes (MSC-Exos), there are intense research investigations to determine how the MSC-Exos could be leverage for clinical applications [4, 56].
MSC-Exos have been associated with anti-inflammatory and reparative functions [55, 57–60]. Over 2000 proteins have been identified within MSC-Exos [54, 55]. These include cytokines (e.g. IL-10, IL-6, TGF-, TNF), chemokines (e.g. MCP1, CXCL14, MCP3, SDF-1), and trophic factors (e.g. FGF, HGF, IGF1, VEGF) [55]. Like their parent cells, MSC-Exos can suppress effector T-cells, dendritic cell (DC) maturation, M1 macrophages, and natural killer cells (NKs), while enhancing regulatory T-cells and M2 macrophages [56, 61–63]. The exosome cargo of MSCs contribute to their regenerative capacity such as enhanced osteogenesis, chondrogenesis, and angiogenesis [60, 64–66]. It should be noted, however, that both direct and indirect forms of intercellular communication between MSCs and target cells have been reported; for example gap junctional intercellular communication (GJIC) for direct intercellular communication, and release of MVs for indirect intercellular communication [67–70]. It should be noted that both means of communication have the potential to transfer similar cargo, including proteins and miRNAs to target cells [71].
MSC Licensing
MSCs can be licensed or educated as anti-inflammatory cells in a tissue niche [14, 72]. While the process of MSC licensing is poorly defined, specific factors are known to be necessary – namely IFN, in the presence of other pro-inflammatory cytokines (e.g. TNF, IL-1, IL-1) [73–75]. The immunomodulatory properties of MSCs were not well understood until recent studies showed that MSCs could impair the function of both innate and adaptive immune system cell proliferation [76, 77]. These studies paved the way for scientists to shift their focus from the MSCs multiple-lineage and regenerative properties towards understanding the immune regulator capacity of these cells. MSCs regulate the immune system response through the release of MVs and soluble factors that impact the ability of the innate and adaptive immune cells such as myeloid DC and T-cells to respond to an infection [73, 78, 79]. MSCs impair DC function by preventing their transition from immature to mature DCs, thus preventing presentation of antigen to naïve T-lymphocytes and decreased release of pro-inflammatory cytokines (e.g. TNF) [79, 80]. MSCs also decreased NK cytotoxicity. This could occur by decreased IFN, leading to reduced NK proliferation, in part through downregulated expression of NKp30 and NKG2D on NK cells [73, 81]. MSCs further regulate the immune system by impairing the inflammatory response induced by the cells of the adaptive immune system. For example, MSCs inhibit T-lymphocytes differentiation by increasing IL-4 and decreasing IFN secretion [73]. The decrease of pro-inflammatory cytokines allows T-lymphocytes to differentiate towards anti-inflammatory T2 rather than pro-inflammatory T1 phenotype [40, 73, 82]. MSCs also act on macrophages by polarization from pro-inflammatory M1 to anti-inflammatory M2 phenotype [61, 83, 84]. These findings have relevance to MSC function in the uterus, as MSCs isolated from and menstrual fluid have immunomodulatory properties [85]. Thus, it has been postulated, although unproven, that similar interactions between MSCs and uterine immune cells (e.g. uterine NK (uNK) cells and macrophages) play a role in promoting the immune microenvironment required for endometrial regeneration [86].
Regenerative Potential of MSCs
MSCs, in addition to their crucial role in immune modulation, also play an essential role in maintaining the host tissues’ homeostasis by replacing dead and dysfunctional tissue. The capacity of MSCs to sustain host homeostasis through repair and replacement of dead and dysfunctional cells is predominantly attributed to their secretome and the microenvironment, as discussed above. The effectiveness of MSCs response to inflammation or injury is conditioned by their ability to home to the site of insult or injury. However, once MSCs are recruited to the site of inflammation, they can directly or indirectly interact with the affected tissue by GJIC or paracrine factors, as indicated above. Local inflammatory cytokines (e.g. TNF, IFN) signal MSCs to release immunomodulatory, pro-angiogenic, regenerative, and neuroprotective factors including TGF-1, VEGF, HGF, SDF-1, IGF-1, and angi-1 [40, 59, 87, 88]. These effects can be accomplished via the MSCs’ endogenous cargo or can be engineered to enhance these effects [1, 2, 89, 90].
A key role for MSCs involves research studies to apply MSCs as a drug delivery tool such as gene therapy [70, 91–93]. The self-renewing capacity of MSCs permits these stem cells to act as self-maintaining drug delivery vehicles at a site of inflammation so long as the microenvironment remains permissive to the MSCs [40, 94, 95]. An advantage of using MSCs in drug delivery is their unique ability to be delivered across the immune barrier. This makes these cells available as off the shelf for immediate use to patients.
Although the research predominantly supports MSCs’ effects on their secreted secretome rather than cellular replacement, the latter could be possible. MSCs have been found to generate osteocytes, adipocytes, chondrocytes, myocytes, and functional neurons in vitro [2, 12, 13, 96, 97]. Numerous groups are investigating ways to accomplish similar direct reprogramming of MSCs and other cells in vivo, for example differentiating fibroblasts into neurons [98]. In order to dissect these two options, it is required to understand the behavior of MSCs within different inflammatory milieu, which is addressed in this review. A key factor in understanding the different inflammatory milieu is the transcription factor nuclear factor B (NFB) due to is role in cytokine regulation as well as other inflammatory mediators.
Nuclear Factor B (NFB) Family and Activation
NFB is a family of constitutively expressed, inducible transcription factors that regulate immune and inflammatory responses by controlling the expression of other genes [99, 100]. NFB is found in the cytoplasm as a homodimer or heterodimer composed of combinations of the family’s five subunits: NFB1 (precursor: p105, mature: p50), NFB2 (precursor: p100, mature: p52), RelA (p65), RelB, and c-Rel (Figure 1) [99, 100]. The NFB dimers then bind the B enhancer element on target genes to mediate gene transcription for various cell processes [101]. Although the predominant binding sequence has been identified as GGRRNNYYCC, alternative, noncanonical binding sites have been identified [102–104]. Under baseline conditions, the NFB proteins are sequestered in the cytoplasm by the ankyrin repeats region of either a member of the IB family of inhibitory proteins (namely IB) or an elongated precursor form of NFB1 or NFB2 [105, 106].
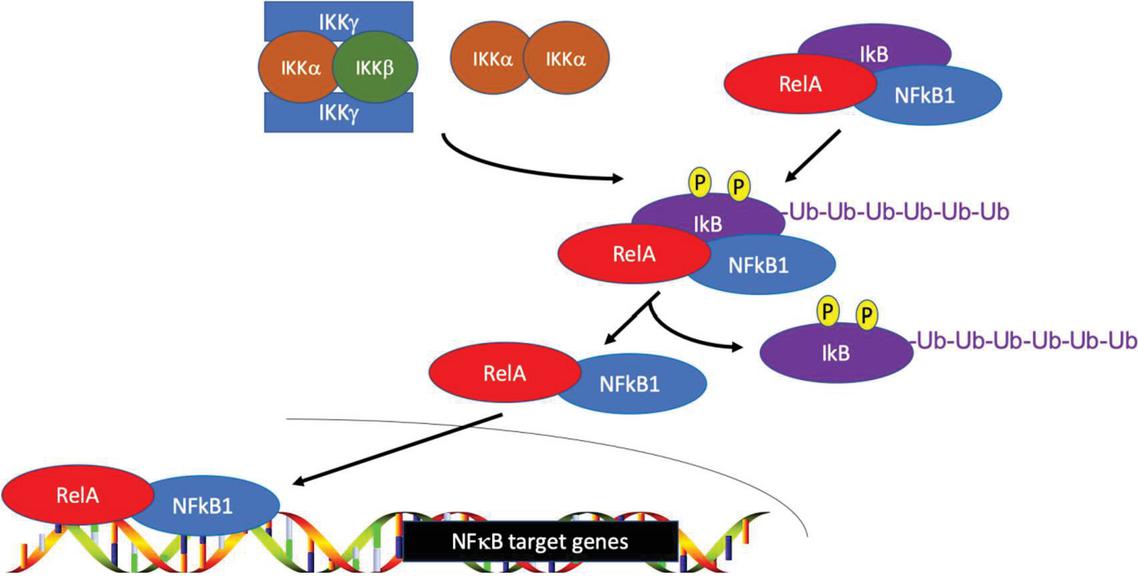
Figure 1 NFB activation. NFB dimers are inactive in the cytoplasm. This occurs by interaction with a IB molecule (or otherwise include an elongated precursor NFB1 or NFB2 subunit). Upon activation, IB (or NFB1 or NFB2) is phosphorylated, which signals ubiquitination, leading to release of the NFB dimer. NFB is then able to translocate to the nucleus, where it can bind its target genes for transcription.
The IB family is composed of seven molecules (IB-, IB-, IB-, IB, Bcl-3, NFB1, and NFB2), which bind the NFB dimer in the cytosol to maintain NFB inactivity (Figure 1). An additional IK member, IB, can bind the NFB dimer in the nucleus. Each of these proteins contains ankyrin repeats, which interact with NFB’s RHD domain inactive [107, 108]. Upon receipt of an NFB activation signal the IB molecule dissociates from the NFB dimer and degrades, permitting NFB translocation to the nucleus. In the case of NFB1 and NFB2, there is cleavage from p105 to p50 and p100 to p52, respectively [107, 108]. Thus, NFB activity can be linked to IB transducing upstream cytoplasmic signals. However, it should also be noted that the IB proteins can be regulated through NFB synthesis, permitting an auto-regulatory NFB feedback loop [109, 110].
NFB activation can be divided into two major signaling pathways: canonical and noncanonical pathways (Figure 2). Although both signaling cascades play important roles in regulating immune and inflammatory responses, the signaling mechanisms have unique upstream activating receptors and signaling molecules [101, 105, 106]. Canonical NFB signaling is initiated by diverse stimuli, including the ligands for various cytokines, pattern recognition receptors, TNF receptor superfamily members, T cell receptors, and B cell receptors; it is primarily activated by toll-like receptors, IL-1, and TNF [99, 111–114]. Through various downstream pathways, these canonical signaling cascades predominantly converge at the inducible degradation of IB: a multi-subunit IB kinase (IKK) phosphorylates IB as specific sites to release the NFB dimer, permitting NFB translocation to the nucleus [99]. The most common NFB dimers observed in canonical NFB signaling are the p50/RelA and p50/c-Rel dimers [99, 106, 115, 116].
IKK itself is composed of two catalytic subunits, IKK and IKK, as a homodimer or heterodimer and its own regulatory subunit NFB essential modulator (NEMO; IKK) [117–119]. IKK can be activated by different inflammatory stimuli, including cytokines, growth factors, mitogens, microbial components, and stress molecules [118, 119]. The activated IKK complex phosphorylates two serines on the N terminus of IB, triggering ubiquitin-dependent degradation of IB by the 26s proteasome and nuclear translocation of the NFB dimer to the nucleus [99, 118].
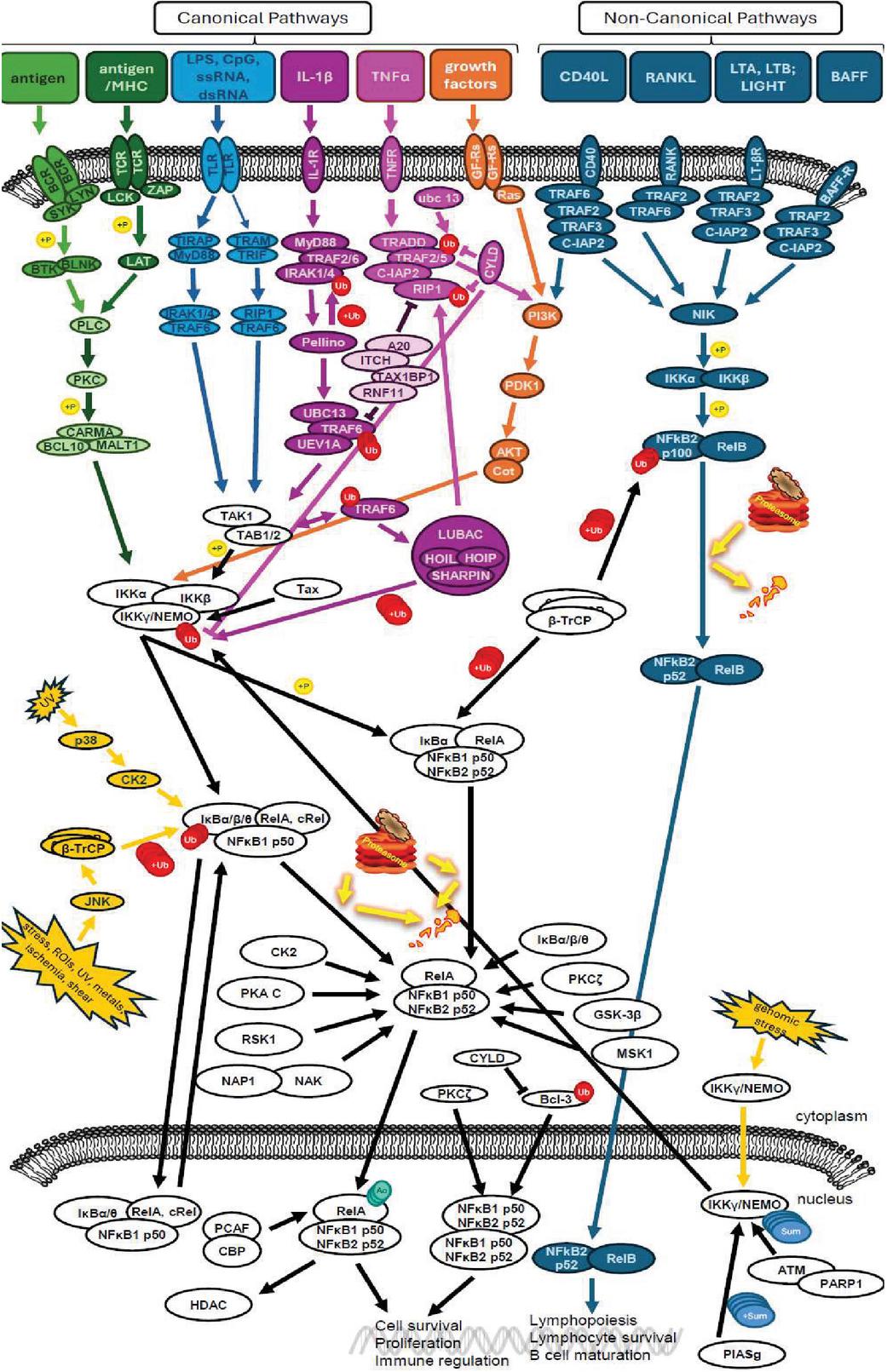
Figure 2 NFB signaling occurs through a complex molecular network with canonical and noncanonical pathways. Shown are overlapping signaling molecules within the pathways such as changes in phosphorylation (p), ubiquitination (UB), sumoylation (Su), and acetylation (Ac). These changes demonstrate influence on downstream NFB transcriptional activity. The figure represents a compilation of the pathways discussed in this article. Some of the components used images from Motifolio and others using PowerPoint.
In noncanonical NFB signaling, activation predominantly occurs through B-cell activation factor, lymphotoxin -receptor, CD40, receptor activator for nuclear factor kappa B (RANK), TNFR2, and fibroblast growth factor-inducible 14 (Fn14) and other ligands of the TNF receptor family [107, 120–123]. These alternative pathways are often associated with lymphoid organogenesis and B-cell function [124, 125]. Rather than inducible degradation of IB, the noncanonical pathways usually converge on processing the NFB2 precursor protein p100 to the mature p52 [105, 121]. p100 undergoes phosphorylation, ubiquitination, and processing through an NFB-inducing kinase (NIK)-IKK complex [124, 126]. Like in the canonical signaling pathways, the ubiquitinated fragment of the protein is processed in the 26s proteasome [124, 126]. This processing yields the mature p52 NFB2 subunit, allowing translocation of the NFB dimer to the nucleus. The most prevalent dimer in non-canonical NFB signaling is p52/RelB [99]. Although both the canonical and noncanonical NFB pathways are involved in almost all aspects of immune responses, the canonical pathway is considered predominant in these functions, with the noncanonical pathways supplementing the canonical pathways and regulating further aspects of adaptive immunity [105, 120].
Targeting NFB in Inflammation and Cancer
NFB is best known for its roles in inflammatory and anti-apoptotic responses [101, 106, 115–117, 120]. NFB induces various pro-inflammatory genes, activates inflammasomes, and regulates the activation and maturation of various immune cells [122, 127, 128]. Depending on the NFB dimer, different target genes, through slight differences in the binding site consensus sequence, are activated including genes linked to inflammation, such as IFN, IL-1, IL-1, IL-10, TNF, TGF-, as well as various growth factors and cell surface receptors [129–134]. The diversity of the NFB binding sequences, in conjunction with the diversity of NFB dimers, makes in silico identification of putative NFB binding sites difficult [104, 135]. Regarding regulation of functions, NFB contributes to a range of innate and adaptive immune functions including regulation of adhesion molecules and apoptotic genes, induction of proliferation genes, and involvement in cell specialization (i.e. Schwann cells myelination) and development [136–141]. Beyond these broad mechanisms, NFB has been implicated in both pro- and anti-inflammation functions.
NFB has been found to activate the expression of cytokines and chemokines, including the pro-inflammatory cytokines TNF and IL-1, which can in turn activate NFB [112–114]. These chemokines activate NFB at different points in time, for different lengths of time, after activating their respective pathways; this can result in biphasic NFB activity [114]. Various chronic inflammatory conditions, in which TNF is elevated, have shown that inhibition of NFB abrogated the elevated TNF with reduction in inflammation [142]. However, mechanisms of blocking NFB have NFB have also resulted in septic shock and inflammation [143–145], suggesting an anti-inflammatory role of NFB as well. However, the full role of NFB in inflammation is yet unclear (reviewed in [146, 147]).
NFB dysregulation has been observed in various inflammatory diseases, including atherosclerosis, inflammatory bowel diseases, multiple sclerosis, and rheumatoid arthritis [99]. Targeting of NFB has been investigated at the levels of IKK inhibition (e.g. aspirin, preventing phosphorylation of IB), proteasome inhibition (e.g. bortezomib, preventing IB degradation), inhibition of nuclear translocation (e.g. tacrolimus), and inhibition of DNA-binding (e.g. glucocorticoids, preventing target gene transcription) (Figure 3) [148–152].
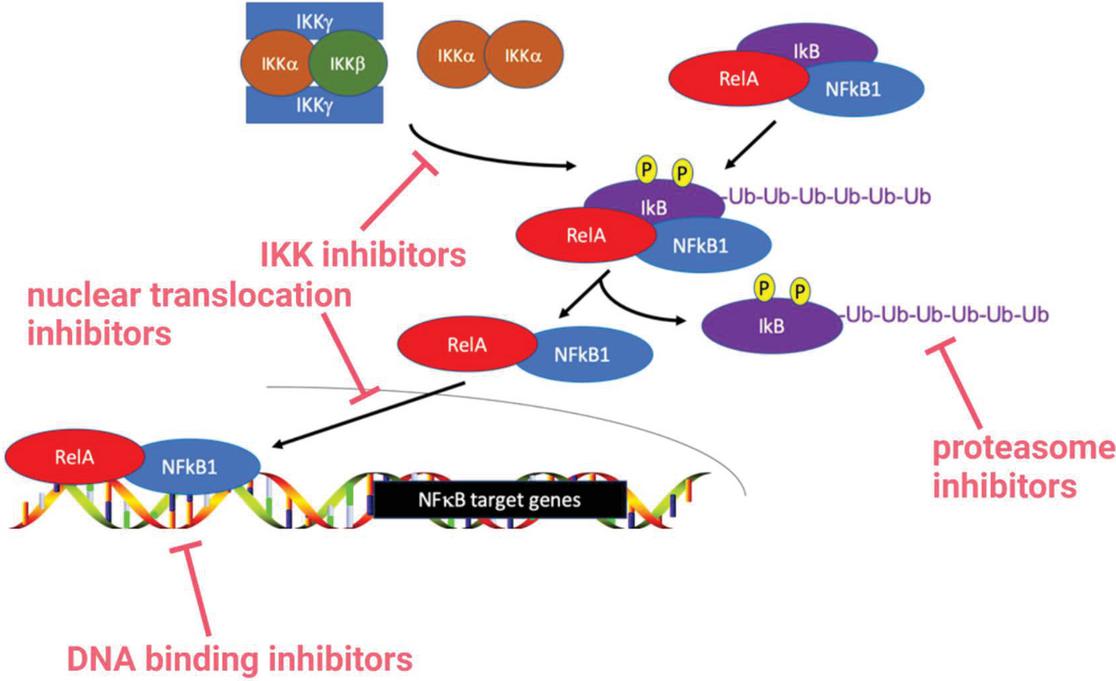
Figure 3 Clinical targeting of NFB. Available pharmacological agents that inhibit NFB at various points, including inhibiting IKK to prevent IB phosphorylation, inhibiting the proteasome to preventing IB degradation, inhibiting nuclear translocation, and inhibiting DNA-binding to prevent target gene transcription.
NFB is among the signaling cascades that are aberrantly regulated in cancer target genes include molecules that are retained intracellularly and extracellularly. This and extracellularly. This allows cancer cells to influence signaling of neighboring non-malignant cells in the microenvironment, resulting in the cancer cells regulating their own cellular support [153–156]. Aberrant NFB signaling has been implicated in all cells of the tumor microenvironment, including tumor-associated macrophages (TAMs), DCs, myeloid-derived suppressor cells (MDSCs), neutrophils, mast cells, NK cells, T-cells, B-cells, cancer-associated fibroblasts (CAFs) and endothelial cells [157].
NFB is involved with all stages of tumor progression: tumor initiation, tumor proliferation, and tumor metastasis [157]. During tumor initiation, NFB can stimulate cell cycle entry of cells containing a single strand of damaged DNA, thus ensuring that a daughter cell has two copies of the damaged gene(s) [158, 159]. Further, NFB can induce transcription of activation-induced cytidine deaminase (AID), a mutator enzyme, which deaminates cytosine residues to cause cytosine-to-thymine transitions [160]. Within the tumor proliferation aspect of tumor progression, NFB can promote cancer cell proliferation and survival through the production of inflammatory cytokines and growth factors [161, 162]. apoptosis that would otherwise be induced by oncogenes like Myc [163]. For tumor metastasis, NFB can induce genes such as SLUG, TWIST, and TRAIL, whose protein products contribute to epithelial mesenchymal transition (EMT) [164, 165]. Additionally, NFB can stimulate the expression of motility factors such as HGF and CXCL12, and HIF1 to enhance survival of the metastatic cells [166–168].
NFB in Stem Cell Multipotency: Healthy and Malignant
Initial studies of NFB in adult stem cells were done in hematopoietic stem cells (HSCs). There is increased NFB activity in HSCs, specifically the RelA subunit that can increase hematopoietic cell proliferation, decreased numbers of long-term HSCs, and increased numbers of short-term HSCs [169–171]. Although increased NFB (RelA) activity led to differentiation of HSCs, the short-term HSCs were unable to reconstitute the hematopoietic system, indicating that these short-term HSCs have lost multipotency [171]. However, further research in MSCs and HSCs have found that NFB maintains multipotency, with NFB inhibiting multilineage differentiation [172–175]. This correlated with a decrease in NFB during differentiation. This was corroborated in studies in which inhibition of NFB initiated MSC differentiation [176, 177]. It has been suggested that the loss of NFB mitigates the proinflammatory microenvironment and thus triggers MSC differentiation [178, 179]. Variable effects on multipotency have been observed when exposing MSCs to NFB-stimulating factors. Specifically, if NFB signaling maintains multipotency of CSCs and MSCs. For example, stimulation with TNF resulted in differentiation of the MSCs while TLR4 did not result in a loss of multipotency [180–182].
NFB is constitutively active in CSCs, stimulating CSC maintenance, proliferation, and expansion [183–185]. Together with STAT3, NFB promotes CSC maintenance through SLUG, TWIST, and SNAIL [183–186]. In breast cancer, NFB has been shown to expand the CSC population by activating Notch signaling [187]. NFB has also been implicated in CSCs from hematological malignancies, including increased NFB activity in AML, CML, and multiple myeloma CSC [188–191]. This elevated NFB activity yields increased expression of stem cell associated genes such as CD44, Sox2, and Nanog within the CSCs [192].
Purinergic Receptors (Purinoceptors)
Purinergic receptors (purinoceptors) are a family of plasma membrane channels and receptors involved in import of adenosine-5-triphosphate (ATP), adenosine-5-diphosphate (ADP), adenosine-5-monophosphate (AMP), and adenosine or their signals into the cell generally associated with inflammatory conditions while adenosine is generally associated with decreased inflammation [194]. This evolutionarily basal system has been evolutionarily basal system has been identified in nearly all mammalian tissues, with roles in embryonic development, the nervous system, and endocrine systems; pain, inflammation and immune repair; and cell proliferation, differentiation, and death, among others [195–198]. During the energy metabolism process, ATP is hydrolyzed into ADP and AMP, followed by adenosine by ectonucleosides [199–201]. In humans, these are predominantly CD39 and CD73; with CD39 among the ectonucleosides responsible for hydrolyzing ATP to ADP and AMP, and CD73 hydrolyzing AMP into adenosine (Figure 4) [199].
Purinoreceptors are divided into three main classes based on their ligand(s) and channel/receptor type: P1 (adenosine, ADORA) receptors, P2X receptors, and P2Y receptors [193, 202]. The P1 (ADORA1/A1R, ADORA2A/A2AR, ADORA2B/A2BR, ADORA3/A3R) receptors are G-protein coupled receptors (GPCRs) selective for adenosine [203]. The P2 receptors are selective for purines and pyrimidines and are divided into P2X and P2Y classes [193, 204]. The P2X receptors (P2X1-7) are ligand-gated ion channels of three homomultimers or heteromultimers selective for ATP [202, 204]. The P2Y receptors (P2Y1/2/4/6/11-14) are GPCRs selective for all nucleotides [202, 204]. The missing P2Y receptor numbers are due to non-human orthologs or non-functional receptors in humans [193].
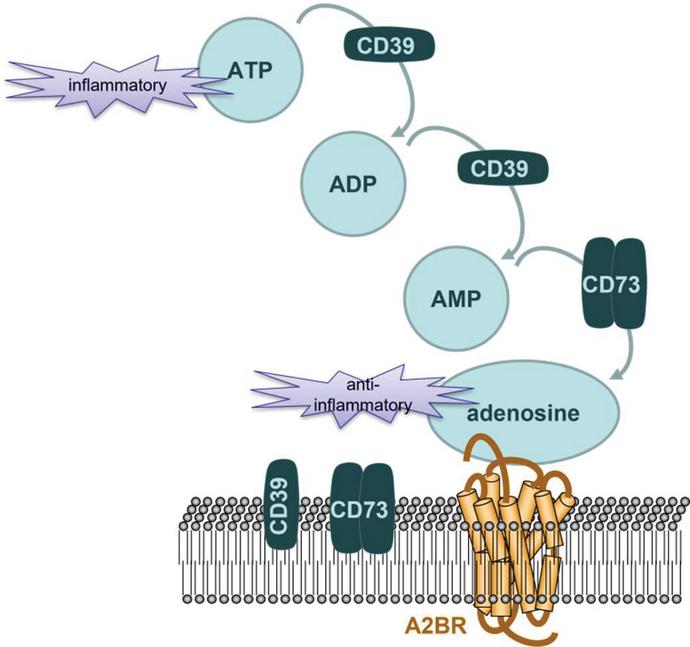
Figure 4 Conversion of ATP to adenosine. ATP is hydrolyzed to ADP, then AMP, by CD39. CD73 then hydrolyzes AMP to adenosine. These metabolic changes from ATP to adenosine can be completed by a cell autonomous method or with the signaling from a neighboring cell. MSCs express both CD39 and CD73 on the cell membrane, permitting them to complete all stages of ATP hydrolysis in a cell-autonomous manner.
Purine-related diseases are complex, involving aberrant purine genesis and metabolism [205–208]. For example, gout is associated with accumulation of urate crystals in joints secondary to a purine-rich diet [209]. Additionally, various tumor microenvironments have been reported to be rich in purinergic receptors. Their substrates, ATP and adenosine, are critical energy signals and storage modulators for tumor metabolism [205, 210–212]. Purine metabolism and purinoceptors have thus become clinical targets, with US Food and Drug Administration (FDA) approved drugs including regadenoson, istradefylline, dipyridamole, clopidogrel, prasugrel, cangrelor, and ticagrelor [205].
The immune system is rich in purinergic receptors and signaling, with almost all immune cells expressing multiple types of purinoceptors [205]. Additionally, CD39 and CD73 are widely expressed on these cells [213, 214]. ATP is generally associated with danger signals and activation of immune cells in which adenosine generally blunts immune responses (Figure 3) [194, 215]. Thus, the P2 receptors are generally considered to be more heavily involved in activation of the immune response while P1 receptors are generally considered more involved in diminishing immune responses [205, 216]. More broadly, purinergic signaling has also been linked to hematopoiesis, including hematopoietic stem cell development and trafficking [217, 218].
Among the P1 adenosine receptors, we focus on ADORA2B due to its association with CD73 through its ligand, adenosine. ADORA1 and ADORA2A have a higher affinity for adenosine than do ADORA2B and ADORA3 [193]. However, ADORA1 and ADORA2A are more closely linked with the central nervous system, while ADORA2B and ADORA3 are more closely associated with immune [219]. Studies agree that ADORA2B is expressed on stem cells, while different groups have reported no or low expression of ADORA3 [220–222]. This variability is seen both for MSCs and for other stem cells, including CSCs. It is possible that this heterogeneity of expression is based on the heterogeneous nature of MSCs in culture, with different labs enhancing different MSC subpopulations as an artifact of culture [2].
ADORA2B is a low abundant protein expressed throughout the body (https://www.proteinatlas.org/ENSG00000170425-ADORA2B) [223]. Its stimulation requires a relatively high concentration of adenosine relative to other purinergic receptors [193]. ADORA2B is involved in various pro- and anti-inflammatory processes [224–226]. Further, changes in ADORA2B activation have been seen with various types of MSC differentiation, most notably increased activation during osteogenic differentiation [227–229]. However, it has also been reported that increased ADORA2B activation inhibits [230]. These differences in ADORA2B activation with respect to differentiation lead to questions regarding the receptor’s role in stem cell multipotency. stem cell multipotency.
Conclusion and Discussion
Globally, there are over six thousand reported clinical trials with MSCs that are ongoing and completed (clinicaltrials.gov). These trials include but are not limited to treatment of immune mediated disorders such as autoimmunity and inflammation, infection, tissue repair and regeneration, and drug delivery [2, 3]. The outcome of these trials are mixed, with several positive outcomes and several positive results. The varied outcomes might be largely due to the combined analyses of results from different sources of MSCs [2, 231]. Specifically, differences in donor and tissue source, method of isolation and expansion, treatments to the MSCs in vitro, and method of delivery. Beyond these differences, much of MSC biology remains unresolved. Notably, the fate of MSCs in vivo is unclear. To date, it is unclear if MSCs can exert functional changes without engrafting long-term [1, 34, 67, 232]. It is clear, however, that MSCs can be engineered to transport drug cargo to target regions (e.g. bone marrow, brain) [2, 40, 70]. MSCs are functionally plastic with the expression of a wide array of cytokines and chemokines, and their receptors, which permit the MSCs to respond rapidly to changing microenvironments [231, 233]. In the presence of an inflammatory milieu, the MSCs are licensed to be immune suppressor cells [72]. Beyond licensing, the fate of the MSCs in vivo is less clear. It is thus critical to understand the microenvironmental interactions to elucidate the fate of MSCs after they release their drug cargo and/or after the cells have mitigated pathologic inflammation.
We and others have reported the role of NFB as a regulator of multipotency [175, 177]. However, it is unclear what NFB pathway is active to maintain multipotency. NFB can activate multiple pathways in MSCs, depending on the inflammatory milieu [234, 235]. There are several studies on the role of cytokines on multipotency of MSCs [236–241]. However, in cell culture the MSCs can maintain multipotency throughout cell passages. This indicated that there must be a cell autonomous method that has not been elucidated, which must be addressed in future studies. In contrast to the overwhelming number of studies on cytokines that can act in a cell-autonomous mechanism to maintain multipotency, there is a family of inflammatory mediators belonging to the purinergic family whose role in multipotency is less understood [242–244]. In this capacity, this review article has highlighted the gap in knowledge by addressing the purinergic signaling in multipotency. Specifically focusing on ADORA2B, a purinergic receptor for adenosine with both pro- and anti-inflammatory functions – a duality shared with MSCs [224–226]. Further, ADORA2B expression is known to change during MSC differentiation, and its expression has been linked with that of NFB [226, 245–247].
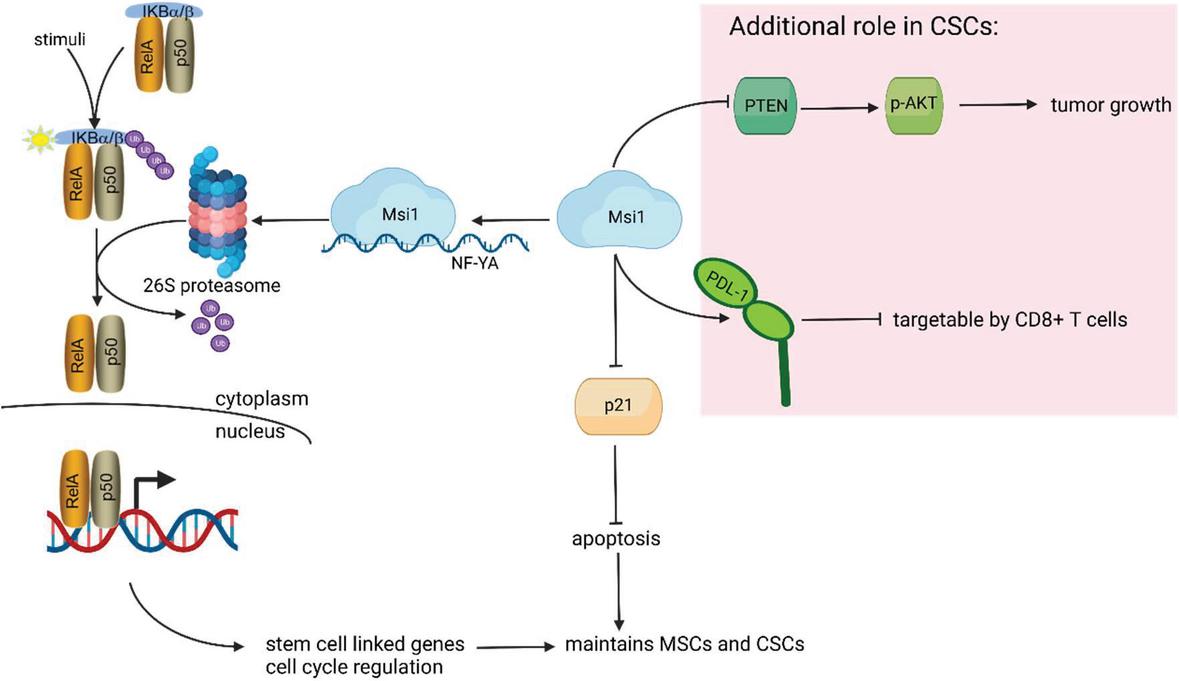
Figure 5 Summary: NFB regulation in stem cell maintenance. RelA-ccontaining NFB complex is sequestered in the cytoplasm by IkB. Through a series of upstream stimuli, IKK phosphorylates IkB, which is subsequently ubiquitinated for degradation by the 26s proteasome. Upon phosphorylation and ubiquitination, IkB releases NFB, which translocates to the nucleus for transcription of stem cell-linked genes. Msi1 induces transcription of NF-YA, a subunit of the 26s proteasome. Msi1 also inhibits p21, thus blocking apoptosis for stem cell maintenance. Specifically in the context of CSCs, Msi1 has the additional functions of blocking PTEN to enhance tumor growth, and increased PD-L1 expression to exert immune suppression such as tumor- targeting CD8 T cells.
The ligand for ADORA2B is adenosine, which requires conversion of ATP to ADP to AMP to adenosine in the extracellular space. The first two steps of this conversion are completed by the ectonucleotidase CD39 (ectonucleoside triphosphate diphosphohydrolase 1); the final conversion of AMP to adenosine is completed by the ectonucleotidases CD73 (5’-nucleotidase ecto) – both of which are expressed on MSCs, among other cells in the microenvironment [201]. CD73 is a constitutive marker for MSCs. However, its role in maintaining the stem cell state is poorly understood [248, 249]. In other cell systems, CD73 has been found to regulate both pro- and anti-inflammatory signaling [247, 250]. In MSCs, it has been suggested that CD73 may be linked to anti-inflammatory function, with CD73 anti-inflammatory function, relative to CD73 MSCs [251]. Additionally, CD73 is upregulated in many cancers, including an observed correlation with increased CD73 and observed correlation with increased CD73 and increased NFB in MSCs derived from patients with myeloproliferative neoplasms [252, 253].
This article discusses a rationale to assign NFB as key to maintaining multipotency of MSCs, partly maintained by cell autonomous process, and exogenously, by the inflammatory milieu. The cell-autonomous method involves the NFB-purinergic axis, specifically the purinoceptor ADORA2B. IKK phosphorylates IkB, which releases the RelA subunit of NFB, permitting nuclear translocation to activate NFB targeted gene transcription (Figure 4). In this regard, inhibition of NFB activity could lead to a loss of multipotency and increased cellular senescence. The experimental inflammatory milieu can activate NFB, directly or indirectly through release of other cytokines such as IL-1, IL-10, TGF- TNF. Activation of NFB, in turn can sustain the high levels of the produced cytokines through autonomous regulation of the MSCs (Figure 4). Stemness discussed in this review is not limited to NFB. In cancer stem cells (CSC), Musashi 1 (Msi1) was shown to maintain CSCs. It appears that Msi1 might be linked to immune checkpoint since PD-L1 was co-expressed with Msi1 on CSCs. Possible mechanism of Msi 1 in CSC maintenance is summarized in Figure 5.
Acknowledgement
The work is in partial fulfillment for a doctoral thesis for LS. The work was supported by New Jersey Commission on Cancer Research.
References
[1] Sherman, L. S., A. Condé-Green, O. A. Sandiford, and P. Rameshwar. 2015. A discussion on adult mesenchymal stem cells for drug delivery: pros and cons. Ther Deliv 6: 1335–1346.
[2] Sherman, L. S., M. Shaker, V. Mariotti, and P. Rameshwar. 2017. Mesenchymal stromal/stem cells in drug therapy: New perspective. Cytotherapy 19: 19–27.
[3] Maldonado, V. V., N. H. Patel, E. E. Smith, C. L. Barnes, M. P. Gustafson, R. R. Rao, and R. M. Samsonraj. 2023. Clinical utility of mesenchymal stem/stromal cells in regenerative medicine and cellular therapy. J Biol Eng 17: 44.
[4] Torrecillas-Baena, B., V. Pulido-Escribano, G. Dorado, M. Gálvez-Moreno, M. Camacho-Cardenosa, and A. Casado-Díaz. 2023. Clinical Potential of Mesenchymal Stem Cell-Derived Exosomes in Bone Regeneration. J Clin Med 12.
[5] Campagnoli, C., I. A. Roberts, S. Kumar, P. R. Bennett, I. Bellantuono, and N. M. Fisk. 2001. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood 98: 2396–2402.
[6] Castillo, M., K. Liu, L. Bonilla, and P. Rameshwar. 2007. The immune properties of mesenchymal stem cells. Int J Biomed Sci 3: 76–80.
[7] Caplan, A. I., and S. P. Bruder. 2001. Mesenchymal stem cells: building blocks for molecular medicine in the 21st century. Trends Mol Med 7: 259–264.
[8] Friedenstein, A. J., K. V. Petrakova, A. I. Kurolesova, and G. P. Frolova. 1968. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 6: 230–247.
[9] Friedenstein, A. J., S. Piatetzky, II, and K. V. Petrakova. 1966. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol 16: 381–390.
[10] Dominici, M., K. Le Blanc, I. Mueller, I. Slaper-Cortenbach, F. Marini, D. Krause, R. Deans, A. Keating, D. Prockop, and E. Horwitz. 2006. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8: 315–317.
[11] Le Blanc, K., C. Tammik, K. Rosendahl, E. Zetterberg, and O. Ringdén. 2003. HLA expression and immunologic propertiesof differentiated and undifferentiated mesenchymal stem cells. Exp Hematol 31: 890–896.
[12] Greco, S. J., C. Zhou, J. H. Ye, and P. Rameshwar. 2007. An interdisciplinary approach and characterization of neuronal cells transdifferentiated from human mesenchymal stem cells. Stem Cells Dev 16: 811–826.
[13] Trzaska, K. A., and P. Rameshwar. 2011. Dopaminergic neuronal differentiation protocol for human mesenchymal stem cells. Methods Mol Biol 698: 295–303.
[14] Potian, J. A., H. Aviv, N. M. Ponzio, J. S. Harrison, and P. Rameshwar. 2003. Veto-like activity of mesenchymal stem cells: functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol 171: 3426–3434.
[15] Patel, S. A., L. Sherman, J. Munoz, and P. Rameshwar. 2008. Immunological properties of mesenchymal stem cells and clinical implications. Arch Immunol Ther Exp (Warsz) 56: 1–8.
[16] Stagg, J., and J. Galipeau. 2013. Mechanisms of immune modulation by mesenchymal stromal cells and clinical translation. Curr Mol Med 13: 856–867.
[17] Sasse, S., A. Skorska, C. A. Lux, G. Steinhoff, R. David, and R. Gaebel. 2019. Angiogenic Potential of Bone Marrow Derived CD133(+) and CD271(+) Intramyocardial Stem Cell Trans- Plantation Post MI. Cells 9: 78.
[18] Conaty, P., L. S. Sherman, Y. Naaldijk, H. Ulrich, A. Stolzing, and P. Rameshwar. 2018. Methods of Mesenchymal Stem Cell Homing to the Blood-Brain Barrier. Methods Mol Biol 1842: 81–91.
[19] Sandiford, O. A., R. J. Donnelly, M. H. El-Far, L. M. Burgmeyer, G. Sinha, S. H. Pamarthi, L. S. Sherman, A. I. Ferrer, D. E. DeVore, S. A. Patel, Y. Naaldijk, S. Alonso, P. Barak, M. Bryan, N. M. Ponzio, R. Narayanan, J. P. Etchegaray, R. Kumar, and P. Rameshwar. 2021. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res 81: 1567–1582.
[20] Mohamed-Ahmed, S., I. Fristad, S. A. Lie, S. Suliman, K. Mustafa, H. Vindenes, and S. B. Idris. 2018. Adipose-derived and bone marrow mesenchymal stem cells: a donor-matched comparison. Stem Cell Res Ther 9: 168.
[21] Romagano, M. P., L. S. Sherman, B. Shadpoor, M. El-Far, S. Souayah, S. H. Pamarthi, J. Kra, A. Hood-Nehra, J. P. Etchegaray, S. F. Williams, and P. Rameshwar. 2022. Aspirin-Mediated Reset of Preeclamptic Placental Stem Cell Transcriptome – Implication for Stabilized Placental Function. Stem Cell Rev Rep 18: 3066–3082.
[22] Sherman, L. S., A. Condé-Green, Y. Naaldijk, E. S. Lee, and P. Rameshwar. 2019. An Enzyme-free Method for Isolation and Expansion of Human Adipose-derived Mesenchymal Stem Cells. J Vis Exp. 154: e59419.
[23] Jung, W. Y., J. H. Kang, K. G. Kim, H. S. Kim, B. I. Jang, Y. H. Park, and I. H. Song. 2015. Human adipose-derived stem cells attenuate inflammatory bowel disease in IL-10 knockout mice. Tissue Cell 47: 86–93.
[24] Kim, Y. S., J. Y. Kim, D. M. Shin, J. W. Huh, S. W. Lee, and Y. M. Oh. 2014. Tracking intravenous adipose-derived mesenchymal stem cells in a model of elastase-induced emphysema. Tuberc Respir Dis (Seoul) 77: 116–123.
[25] Le Blanc, K., C. Tammik, K. Rosendahl, E. Zetterberg, and O. Ringdén. 2003. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol 31: 890–896.
[26] Sherman, L. S., J. Munoz, S. A. Patel, M. A. Dave, I. Paige, and P. Rameshwar. 2011. Moving from the laboratory bench to patients’ bedside: considerations for effective therapy with stem cells. Clin Transl Sci 4: 380–386.
[27] Ponte, A. L., E. Marais, N. Gallay, A. Langonné, B. Delorme, O. Hérault, P. Charbord, and J. Domenech. 2007. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells 25: 1737–1745.
[28] Honczarenko, M., Y. Le, M. Swierkowski, I. Ghiran, A. M. Glodek, and L. E. Silberstein. 2006. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells 24: 1030–1041.
[29] Wynn, R. F., C. A. Hart, C. Corradi-Perini, L. O’Neill, C. A. Evans, J. E. Wraith, L. J. Fairbairn, and I. Bellantuono. 2004. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood 104: 2643–2645.
[30] Vanden Berg-Foels, W. S. 2014. In situ tissue regeneration: chemoattractants for endogenous stem cell recruitment. Tissue Eng Part B Rev 20: 28–39.
[31] Hocking, A. M. 2015. The Role of Chemokines in Mesenchymal Stem Cell Homing to Wounds. Adv Wound Care (New Rochelle) 4: 623–630.
[32] Kim, I., H. Park, I. Hwang, D. Moon, H. Yun, E. J. Lee, and H. S. Kim. 2021. Discovery of chemerin as the new chemoattractant of human mesenchymal stem cells. Cell Biosci 11: 120.
[33] Cornelissen, A. S., M. W. Maijenburg, M. A. Nolte, and C. Voermans. 2015. Organ-specific migration of mesenchymal stromal cells: Who, when, where and why? Immunol Lett 168: 159–169.
[34] Iso, Y., J. L. Spees, C. Serrano, B. Bakondi, R. Pochampally, Y. H. Song, B. E. Sobel, P. Delafontaine, and D. J. Prockop. 2007. Multipotent human stromal cells improve cardiac function after myocardial infarction in mice without long-term engraftment. Biochem Biophys Res Commun 354: 700–706.
[35] Liesveld, J. L., N. Sharma, and O. S. Aljitawi. 2020. Stem cell homing: From physiology to therapeutics. Stem Cells 38: 1241–1253.
[36] Braid, L. R., C. A. Wood, D. M. Wiese, and B. N. Ford. 2018. Intramuscular administration potentiates extended dwell time of mesenchymal stromal cells compared to other routes. Cytotherapy 20: 232–244.
[37] Naderi-Meshkin, H., A. R. Bahrami, H. R. Bidkhori, M. Mirahmadi, and N. Ahmadiankia. 2015. Strategies to improve homing of mesenchymal stem cells for greater efficacy in stem cell therapy. Cell Biol Intl 39: 23–34.
[38] Lee, R. H., A. A. Pulin, M. J. Seo, D. J. Kota, J. Ylostalo, B. L. Larson, L. Semprun-Prieto, P. Delafontaine, and D. J. Prockop. 2009. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 5: 54–63.
[39] Ren, G., X. Zhao, L. Zhang, J. Zhang, A. L’Huillier, W. Ling, A. I. Roberts, A. D. Le, S. Shi, C. Shao, and Y. Shi. 2010. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol 184: 2321–2328.
[40] Sherman, L. S., M. P. Romagano, S. F. Williams, and P. Rameshwar. 2019. Mesenchymal stem cell therapies in brain disease. Semin Cell Dev Biol 95: 111–119.
[41] Soukup, R., I. Gerner, T. Mohr, S. Gueltekin, J. Grillari, and F. Jenner. 2023. Mesenchymal Stem Cell Conditioned Medium Modulates Inflammation in Tenocytes: Complete Conditioned Medium Has Superior Therapeutic Efficacy than Its Extracellular Vesicle Fraction. Int J Mol Sci 24.
[42] Lubis, A. M. T., P. Aprianto, J. A. Pawitan, B. P. Priosoeryanto, T. I. T. Dewi, and A. F. Kamal. 2023. Intra-articular injection of secretome, derived from umbilical cord mesenchymal stem cell, enhances the regeneration process of cartilage in early-stage osteo-arthritis: an animal study. Acta Orthop 94: 300–306.
[43] Fischer, U. M., M. T. Harting, F. Jimenez, W. O. Monzon-Posadas, H. Xue, S. I. Savitz, G. A. Laine, and C. S. Cox, Jr. 2009. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev 18: 683–692.
[44] Kerkelä, E., T. Hakkarainen, T. Mäkelä, M. Raki, O. Kambur, L. Kilpinen, J. Nikkilä, S. Lehtonen, I. Ritamo, R. Pernu, M. Pietilä, R. Takalo, T. Juvonen, K. Bergström, E. Kalso, L. Valmu, S. Laitinen, P. Lehenkari, and J. Nystedt. 2013. Transient proteolytic modification of mesenchymal stromal cells increases lung clearance rate and targeting to injured tissue. Stem Cells Transl Med 2: 510–520.
[45] Moeinabadi-Bidgoli, K., R. Mazloomnejad, A. Beheshti Maal, H. Asadzadeh Aghdaei, M. Kazem Arki, N. Hossein-Khannazer, and M. Vosough. 2023. Genetic modification and preconditioning strategies to enhance functionality of mesenchymal stromal cells: a clinical perspective. Expert Opin Biol Ther 23: 461–478.
[46] Kim, D. S., J. H. Kim, J. K. Lee, S. J. Choi, J. S. Kim, S. S. Jeun, W. Oh, Y. S. Yang, and J. W. Chang. 2009. Overexpression of CXC chemokine receptors is required for the superior glioma-tracking property of umbilical cord blood-derived mesenchymal stem cells. Stem Cells Dev 18: 511–519.
[47] Haider, H., S. Jiang, N. M. Idris, and M. Ashraf. 2008. IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1alpha/CXCR4 signaling to promote myocardial repair. Circ Res 103: 1300–1308.
[48] Huang, B., J. Qian, J. Ma, Z. Huang, Y. Shen, X. Chen, A. Sun, J. Ge, and H. Chen. 2014. Myocardial transfection of hypoxia-inducible factor-1 and co-transplantation of mesenchymal stem cells enhance cardiac repair in rats with experimental myocardial infarction. Stem Cell Res Ther 5: 22.
[49] Vittorio, O., P. Quaranta, V. Raffa, N. Funel, D. Campani, S. Pelliccioni, B. Longoni, F. Mosca, A. Pietrabissa, and A. Cuschieri. 2011. Magnetic carbon nanotubes: a new tool for shepherding mesenchymal stem cells by magnetic fields. Nanomedicine 6: 43–54.
[50] Doyle, L. M., and M. Z. Wang. 2019. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 8: 727.
[51] Zhou, H., T. Pisitkun, A. Aponte, P. S. Yuen, J. D. Hoffert, H. Yasuda, X. Hu, L. Chawla, R. F. Shen, M. A. Knepper, and R. A. Star. 2006. Exosomal Fetuin-A identified by proteomics: a novel urinary biomarker for detecting acute kidney injury. Kidney Int 70: 1847–1857.
[52] Sandfeld-Paulsen, B., N. Aggerholm-Pedersen, R. Bæk, K. R. Jakobsen, P. Meldgaard, B. H. Folkersen, T. R. Rasmussen, K. Varming, M. M. Jørgensen, and B. S. Sorensen. 2016. Exosomal proteins as prognostic biomarkers in non-small cell lung cancer. Mol Oncol 10: 1595–1602.
[53] Simpson, R. J., J. W. Lim, R. L. Moritz, and S. Mathivanan. 2009. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics 6: 267–283.
[54] Deng, H., C. Sun, Y. Sun, H. Li, L. Yang, D. Wu, Q. Gao, and X. Jiang. 2018. Lipid, Protein, and MicroRNA Composition Within Mesenchymal Stem Cell-Derived Exosomes. Cell Reprogram 20: 178–186.
[55] Harrell, C. R., N. Jovicic, V. Djonov, and V. Volarevic. 2020. Therapeutic Use of Mesenchymal Stem Cell-Derived Exosomes: From Basic Science to Clinics. Pharmaceutics 12: 474.
[56] Lee, B. C., I. Kang, and K. R. Yu. 2021. Therapeutic Features and Updated Clinical Trials of Mesenchymal Stem Cell (MSC)-Derived Exosomes. J Clin Med 10.
[57] Reis, L. A., F. T. Borges, M. J. Simões, A. A. Borges, R. Sinigaglia-Coimbra, and N. Schor. 2012. Bone marrow-derived mesenchymal stem cells repaired but did not prevent gentamicin-induced acute kidney injury through paracrine effects in rats. PLoS One 7: e44092.
[58] Gatti, S., S. Bruno, M. C. Deregibus, A. Sordi, V. Cantaluppi, C. Tetta, and G. Camussi. 2011. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant 26: 1474–1483.
[59] Allahdadi, K. J., T. A. de Santana, G. C. Santos, C. M. Azevedo, R. A. Mota, C. K. Nonaka, D. N. Silva, C. X. R. Valim, C. P. Figueira, W. L. C. Dos Santos, R. F. do Espirito Santo, A. F. Evangelista, C. F. Villarreal, R. R. Dos Santos, B. S. F. de Souza, and M. B. P. Soares. 2019. IGF-1 overexpression improves mesenchymal stem cell survival and promotes neurological recovery after spinal cord injury. Stem Cell Res Ther 10: 146.
[60] Ren, S., Y. Lin, W. Liu, L. Yang, and M. Zhao. 2023. MSC-Exos: Important active factor of bone regeneration. Front Bioeng Biotechnol 11: 1136453.
[61] Willis, G. R., A. Fernandez-Gonzalez, J. Anastas, S. H. Vitali, X. Liu, M. Ericsson, A. Kwong, S. A. Mitsialis, and S. Kourembanas. 2018. Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation. Am J Respir Crit Care Med 197: 104–116.
[62] Sotiropoulou, P. A., S. A. Perez, A. D. Gritzapis, C. N. Baxevanis, and M. Papamichail. 2006. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 24: 74–85.
[63] He, X., Z. Dong, Y. Cao, H. Wang, S. Liu, L. Liao, Y. Jin, L. Yuan, and B. Li. 2019. MSC-Derived Exosome Promotes M2 Polarization and Enhances Cutaneous Wound Healing. Stem Cells Int 2019: 7132708.
[64] Liang, X., L. Zhang, S. Wang, Q. Han, and R. C. Zhao. 2016. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J Cell Sci 129: 2182–2189.
[65] Jia, Y., S. Qiu, J. Xu, Q. Kang, and Y. Chai. 2020. Exosomes Secreted by Young Mesenchymal Stem Cells Promote New Bone Formation During Distraction Osteogenesis in Older Rats. Calcif Tissue Int 106: 509–517.
[66] Zhang, S., S. J. Chuah, R. C. Lai, J. H. P. Hui, S. K. Lim, and W. S. Toh. 2018. MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials 156: 16–27.
[67] Arslan, F., R. C. Lai, M. B. Smeets, L. Akeroyd, A. Choo, E. N. Aguor, L. Timmers, H. V. van Rijen, P. A. Doevendans, G. Pasterkamp, S. K. Lim, and D. P. de Kleijn. 2013. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res 10: 301–312.
[68] Lim, P. K., S. A. Bliss, S. A. Patel, M. Taborga, M. A. Dave, L. A. Gregory, S. J. Greco, M. Bryan, P. S. Patel, and P. Rameshwar. 2011. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res 71: 1550–1560.
[69] Chen, J., C. Li, and L. Chen. 2015. The Role of Microvesicles Derived from Mesenchymal Stem Cells in Lung Diseases. Biomed Res Int 2015: 985814.
[70] Bliss, S. A., G. Sinha, O. A. Sandiford, L. M. Williams, D. J. Engelberth, K. Guiro, L. L. Isenalumhe, S. J. Greco, S. Ayer, and M. Bryan. 2016. Mesenchymal stem cell–derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res 76: 5832–5844.
[71] Valadi, H., K. Ekström, A. Bossios, M. Sjöstrand, J. J. Lee, and J. O. Lötvall. 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9: 654–659.
[72] Kang, H. S., M. Habib, J. Chan, C. Abavana, J. A. Potian, N. M. Ponzio, and P. Rameshwar. 2005. A paradoxical role for IFN-gamma in the immune properties of mesenchymal stem cells during viral challenge. Exp Hematol 33: 796–803.
[73] Aggarwal, S., and M. F. Pittenger. 2005. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 105: 1815–1822.
[74] Ren, G., L. Zhang, X. Zhao, G. Xu, Y. Zhang, A. I. Roberts, R. C. Zhao, and Y. Shi. 2008. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2: 141–150.
[75] Valencic, E., C. Loganes, S. Cesana, E. Piscianz, G. Gaipa, E. Biagi, and A. Tommasini. 2014. Inhibition of mesenchymal stromal cells by pre-activated lymphocytes and their culture media. Stem Cell Res Ther 5: 3.
[76] Di Nicola, M., C. Carlo-Stella, M. Magni, M. Milanesi, P. D. Longoni, P. Matteucci, S. Grisanti, and A. M. Gianni. 2002. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 99: 3838–3843.
[77] Li, Y. P., S. Paczesny, E. Lauret, S. Poirault, P. Bordigoni, F. Mekhloufi, O. Hequet, Y. Bertrand, J. P. Ou-Yang, J. F. Stoltz, P. Miossec, and A. Eljaafari. 2008. Human mesenchymal stem cells license adult CD34+ hemopoietic progenitor cells to differentiate into regulatory dendritic cells through activation of the Notch pathway. J Immunol 180: 1598–1608.
[78] Uccelli, A., L. Moretta, and V. Pistoia. 2008. Mesenchymal stem cells in health and disease. Nature Reviews Immunology 8: 726–736.
[79] Jiang, X. X., Y. Zhang, B. Liu, S. X. Zhang, Y. Wu, X. D. Yu, and N. Mao. 2005. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood 105: 4120–4126.
[80] Aiello, A., F. Farzaneh, G. Candore, C. Caruso, S. Davinelli, C. M. Gambino, M. E. Ligotti, N. Zareian, and G. Accardi. 2019. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front Immunol 10: 2247.
[81] Spaggiari, G. M., A. Capobianco, S. Becchetti, M. C. Mingari, and L. Moretta. 2006. Mesenchymal stem cell-natural killer cell interactions: evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 107: 1484–1490.
[82] Bai, L., D. P. Lennon, V. Eaton, K. Maier, A. I. Caplan, S. D. Miller, and R. H. Miller. 2009. Human bone marrow-derived mesenchymal stem cells induce Th2-polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia 57: 1192–1203.
[83] Luz-Crawford, P., F. Djouad, K. Toupet, C. Bony, M. Franquesa, M. J. Hoogduijn, C. Jorgensen, and D. Noël. 2016. Mesenchymal Stem Cell-Derived Interleukin 1 Receptor Antagonist Promotes Macrophage Polarization and Inhibits B Cell Differentiation. Stem Cells 34: 483–492.
[84] Lu, D., X. Jiao, W. Jiang, L. Yang, Q. Gong, X. Wang, M. Wei, and S. Gong. 2023. Mesenchymal stem cells influence monocyte/macrophage phenotype: Regulatory mode and potential clinical applications. Biomed Pharmacother 165: 115042.
[85] Bozorgmehr, M., S. Gurung, S. Darzi, S. Nikoo, S. Kazemnejad, A. H. Zarnani, and C. E. Gargett. 2020. Endometrial and Menstrual Blood Mesenchymal Stem/Stromal Cells: Biological Properties and Clinical Application. Front Cell Dev Biol 8: 497.
[86] Salamonsen, L. A., J. C. Hutchison, and C. E. Gargett. 2021. Cyclical endometrial repair and regeneration. Development 148: dev199577.
[87] Gjorgieva, D., N. Zaidman, and D. Bosnakovski. 2013. Mesenchymal stem cells for anti-cancer drug delivery. Recent Pat Anticancer Drug Discov 8: 310–318.
[88] Wang, X., H. Jiang, L. Guo, S. Wang, W. Cheng, L. Wan, Z. Zhang, L. Xing, Q. Zhou, X. Yang, H. Han, X. Chen, and X. Wu. 2021. SDF-1 secreted by mesenchymal stem cells promotes the migration of endothelial progenitor cells via CXCR4/PI3K/AKT pathway. J Mol Histol 52: 1155–1164.
[89] Niu, J., W. Yue, Z. Le-Le, L. Bin, and X. Hu. 2017. Mesenchymal stem cells inhibit T cell activation by releasing TGF-1 from TGF-1/GARP complex. Oncotarget 8: 99784–99800.
[90] Shigemoto-Kuroda, T., J. Y. Oh, D. K. Kim, H. J. Jeong, S. Y. Park, H. J. Lee, J. W. Park, T. W. Kim, S. Y. An, D. J. Prockop, and R. H. Lee. 2017. MSC-derived Extracellular Vesicles Attenuate Immune Responses in Two Autoimmune Murine Models: Type 1 Diabetes and Uveoretinitis. Stem Cell Reports 8: 1214–1225.
[91] Greco, S. J., and P. Rameshwar. 2012. Mesenchymal stem cells in drug/gene delivery: implications for cell therapy. Ther Deliv 3: 997–1004.
[92] Munoz, J. L., S. A. Bliss, S. J. Greco, S. H. Ramkissoon, K. L. Ligon, and P. Rameshwar. 2013. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol Ther Nucleic Acids 2: e126.
[93] Shah, K. 2012. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev 64: 739–748.
[94] Lin, P., Y. Lin, D. P. Lennon, D. Correa, M. Schluchter, and A. I. Caplan. 2012. Efficient lentiviral transduction of human mesenchymal stem cells that preserves proliferation and differentiation capabilities. Stem Cells Transl Med 1: 886–897.
[95] Loebinger, M. R., A. Eddaoudi, D. Davies, and S. M. Janes. 2009. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res 69: 4134–4142.
[96] Rohban, R., and T. R. Pieber. 2017. Mesenchymal Stem and Progenitor Cells in Regeneration: Tissue Specificity and Regenerative Potential. Stem Cells Int 2017: 5173732.
[97] Cho, K. J., K. A. Trzaska, S. J. Greco, J. McArdle, F. S. Wang, J. H. Ye, and P. Rameshwar. 2005. Neurons derived from human mesenchymal stem cells show synaptic transmission and can be induced to produce the neurotransmitter substance P by interleukin-1 alpha. Stem Cells 23: 383–391.
[98] Torper, O., U. Pfisterer, D. A. Wolf, M. Pereira, S. Lau, J. Jakobsson, A. Björklund, S. Grealish, and M. Parmar. 2013. Generation of induced neurons via direct conversion in vivo. Proc Natl Acad Sci U S A 110: 7038–7043.
[99] Liu, T., L. Zhang, D. Joo, and S. C. Sun. 2017. NF-B signaling in inflammation. Signal Transduct Target Ther 2: 1–9.
[100] Oeckinghaus, A., and S. Ghosh. 2009. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1: a000034.
[101] Sun, S. C., J. H. Chang, and J. Jin. 2013. Regulation of nuclear factor-B in autoimmunity. Trends Immunol 34: 282–289.
[102] Linnell, J., R. Mott, S. Field, D. P. Kwiatkowski, J. Ragoussis, and I. A. Udalova. 2004. Quantitative high-throughput analysis of transcription factor binding specificities. Nucleic Acids Res 32: e44.
[103] Chen, F. E., D. B. Huang, Y. Q. Chen, and G. Ghosh. 1998. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 391: 410–413.
[104] Wong, D., A. Teixeira, S. Oikonomopoulos, P. Humburg, I. N. Lone, D. Saliba, T. Siggers, M. Bulyk, D. Angelov, S. Dimitrov, I. A. Udalova, and J. Ragoussis. 2011. Extensive characterization of NF-kappaB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol 12: R70.
[105] Sun, S. C. 2011. Non-canonical NF-B signaling pathway. Cell Res 21: 71–85.
[106] Beinke, S., and S. C. Ley. 2004. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J 382: 393–409.
[107] Hoesel, B., and J. A. Schmid. 2013. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 12: 86.
[108] Hatada, E. N., A. Nieters, F. G. Wulczyn, M. Naumann, R. Meyer, G. Nucifora, T. W. McKeithan, and C. Scheidereit. 1992. The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc Natl Acad Sci U S A 89: 2489–2493.
[109] Sun, S. C., P. A. Ganchi, D. W. Ballard, and W. C. Greene. 1993. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 259: 1912–1915.
[110] Scott, M. L., T. Fujita, H. C. Liou, G. P. Nolan, and D. Baltimore. 1993. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev 7: 1266–1276.
[111] Zhang, H., and S. C. Sun. 2015. NF-B in inflammation and renal diseases. Cell Biosci 5: 63.
[112] Zuckerman, S. H., G. F. Evans, and L. Guthrie. 1991. Transcriptional and post-transcriptional mechanisms involved in the differential expression of LPS-induced IL-1 and TNF mRNA. Immunol 73: 460–465.
[113] Foxwell, B. M., J. Bondeson, F. Brennan, and M. Feldmann. 2000. Adenoviral transgene delivery provides an approach to identifying important molecular processes in inflammation: evidence for heterogenecity in the requirement for NFkappaB in tumour necrosis factor production. Ann Rheum Dis 59 Suppl 1: i54–59.
[114] Johnson, D. R., I. Douglas, A. Jahnke, S. Ghosh, and J. S. Pober. 1996. A sustained reduction in IkappaB-beta may contribute to persistent NF-kappaB activation in human endothelial cells. J Biol Chem 271: 16317–16322.
[115] Karin, M., and M. Delhase. 2000. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin Immunol 12: 85–98.
[116] Hayden, M. S., and S. Ghosh. 2008. Shared principles in NF-kappaB signaling. Cell 132: 344–362.
[117] Sun, S. C., and S. C. Ley. 2008. New insights into NF-kappaB regulation and function. Trends Immunol 29: 469–478.
[118] Israël, A. 2010. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol 2: a000158.
[119] O’Mahony, A., X. Lin, R. Geleziunas, and W. C. Greene. 2000. Activation of the heterodimeric IkappaB kinase alpha (IKKalpha)-IKKbeta complex is directional: IKKalpha regulates IKKbeta under both basal and stimulated conditions. Mol Cell Biol 20: 1170–1178.
[120] Sun, S. C., and Z. G. Liu. 2011. A special issue on NF-B signaling and function. Cell Res 21: 1–2.
[121] Sun, S. C. 2012. The noncanonical NF-B pathway. Immunol Rev 246: 125–140.
[122] Lawrence, T. 2009. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1: a001651.
[123] Sun, S. C. 2011. Non-canonical NF-kappaB signaling pathway. Cell Res 21: 71–85.
[124] Senftleben, U., Y. Cao, G. Xiao, F. R. Greten, G. Krähn, G. Bonizzi, Y. Chen, Y. Hu, A. Fong, S. C. Sun, and M. Karin. 2001. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293: 1495–1499.
[125] Bonizzi, G., M. Bebien, D. C. Otero, K. E. Johnson-Vroom, Y. Cao, D. Vu, A. G. Jegga, B. J. Aronow, G. Ghosh, R. C. Rickert, and M. Karin. 2004. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J 23: 4202–4210.
[126] Xiao, G., E. W. Harhaj, and S. C. Sun. 2001. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell 7: 401–409.
[127] Tak, P. P., and G. S. Firestein. 2001. NF-kappaB: a key role in inflammatory diseases. J Clin Invest 107: 7–11.
[128] Sutterwala, F. S., S. Haasken, and S. L. Cassel. 2014. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319: 82–95.
[129] Sica, A., T. H. Tan, N. Rice, M. Kretzschmar, P. Ghosh, and H. A. Young. 1992. The c-rel protooncogene product c-Rel but not NF-kappa B binds to the intronic region of the human interferon-gamma gene at a site related to an interferon-stimulable response element. Proc Natl Acad Sci U S A 89: 1740–1744.
[130] Mori, N., and D. Prager. 1996. Transactivation of the interleukin-1alpha promoter by human T-cell leukemia virus type I and type II Tax proteins. Blood 87: 3410–3417.
[131] Hiscott, J., J. Marois, J. Garoufalis, M. D’Addario, A. Roulston, I. Kwan, N. Pepin, J. Lacoste, H. Nguyen, G. Bensi, and et al. 1993. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol 13: 6231–6240.
[132] Xu, L. G., and H. B. Shu. 2002. TNFR-associated factor-3 is associated with BAFF-R and negatively regulates BAFF-R-mediated NF-kappa B activation and IL-10 production. J Immunol 169: 6883–6889.
[133] Collart, M. A., P. Baeuerle, and P. Vassalli. 1990. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 10: 1498–1506.
[134] Messer, G., E. H. Weiss, and P. A. Baeuerle. 1990. Tumor necrosis factor beta (TNF-beta) induces binding of the NF-kappa B transcription factor to a high-affinity kappa B element in the TNF-beta promoter. Cytokine 2: 389–397.
[135] Smale, S. T. 2011. Hierarchies of NF-kappaB target-gene regulation. Nat Immunol 12: 689–694.
[136] Collins, T., M. A. Read, A. S. Neish, M. Z. Whitley, D. Thanos, and T. Maniatis. 1995. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J 9: 899–909.
[137] Duckett, C. S. 2002. Apoptosis and NF-kappa B: the FADD connection. J Clin Invest 109: 579–580.
[138] Guttridge, D. C., C. Albanese, J. Y. Reuther, R. G. Pestell, and A. S. Baldwin, Jr. 1999. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 19: 5785–5799.
[139] La Rosa, F. A., J. W. Pierce, and G. E. Sonenshein. 1994. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol Cell Biol 14: 1039–1044.
[140] Nickols, J. C., W. Valentine, S. Kanwal, and B. D. Carter. 2003. Activation of the transcription factor NF-kappaB in Schwann cells is required for peripheral myelin formation. Nat Neurosci 6: 161–167.
[141] Snapper, C. M., P. Zelazowski, F. R. Rosas, M. R. Kehry, M. Tian, D. Baltimore, and W. C. Sha. 1996. B cells from p50/NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol 156: 183–191.
[142] Neurath, M. F., S. Pettersson, K. H. Meyer zum Buschenfelde, and W. Strober. 1996. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med 2: 998–1004.
[143] Nenci, A., C. Becker, A. Wullaert, R. Gareus, G. van Loo, S. Danese, M. Huth, A. Nikolaev, C. Neufert, B. Madison, D. Gumucio, M. F. Neurath, and M. Pasparakis. 2007. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446: 557–561.
[144] Greten, F. R., M. C. Arkan, J. Bollrath, L. C. Hsu, J. Goode, C. Miething, S. I. Goktuna, M. Neuenhahn, J. Fierer, S. Paxian, N. Van Rooijen, Y. Xu, T. O’Cain, B. B. Jaffee, D. H. Busch, J. Duyster, R. M. Schmid, L. Eckmann, and M. Karin. 2007. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130: 918–931.
[145] Bruey, J. M., N. Bruey-Sedano, F. Luciano, D. Zhai, R. Balpai, C. Xu, C. L. Kress, B. Bailly-Maitre, X. Li, A. Osterman, S. Matsuzawa, A. V. Terskikh, B. Faustin, and J. C. Reed. 2007. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129: 45–56.
[146] Ben-Neriah, Y., and M. Karin. 2011. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 12: 715–723.
[147] Wang, Y., X. Chen, W. Cao, and Y. Shi. 2014. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol 15: 1009–1016.
[148] Lin, Y., L. Bai, W. Chen, and S. Xu. 2010. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets 14: 45–55.
[149] Yin, M. J., Y. Yamamoto, and R. B. Gaynor. 1998. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396: 77–80.
[150] Veschini, L., D. Belloni, C. Foglieni, M. G. Cangi, M. Ferrarini, F. Caligaris-Cappio, and E. Ferrero. 2007. Hypoxia-inducible transcription factor-1 alpha determines sensitivity of endothelial cells to the proteosome inhibitor bortezomib. Blood 109: 2565–2570.
[151] Jennings, C., B. Kusler, and P. P. Jones. 2009. Calcineurin inactivation leads to decreased responsiveness to LPS in macrophages and dendritic cells and protects against LPS-induced toxicity in vivo. Innate Immunol 15: 109–120.
[152] Granelli-Piperno, A., P. Nolan, K. Inaba, and R. M. Steinman. 1990. The effect of immunosuppressive agents on the induction of nuclear factors that bind to sites on the interleukin 2 promoter. J Exp Med 172: 1869–1872.
[153] Taniguchi, K., and M. Karin. 2018. NF-B, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18: 309–324.
[154] Kong, X., L. Li, Z. Li, and K. Xie. 2012. Targeted destruction of the orchestration of the pancreatic stroma and tumor cells in pancreatic cancer cases: molecular basis for therapeutic implications. Cytokine Growth Factor Rev 23: 343–356.
[155] Sionov, R. V., Z. G. Fridlender, and Z. Granot. 2015. The Multifaceted Roles Neutrophils Play in the Tumor Microenvironment. Cancer Microenviron 8: 125–158.
[156] Bonavita, E., M. R. Galdiero, S. Jaillon, and A. Mantovani. 2015. Phagocytes as Corrupted Policemen in Cancer-Related Inflammation. Adv Cancer Res 128: 141–171.
[157] Taniguchi, K., and M. Karin. 2018. NF-B, inflammation, immunity and cancer: coming of age. Nature Rev Immunol 18: 309–324.
[158] Joyce, D., C. Albanese, J. Steer, M. Fu, B. Bouzahzah, and R. G. Pestell. 2001. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev 12: 73–90.
[159] Kiraly, O., G. Gong, W. Olipitz, S. Muthupalani, and B. P. Engelward. 2015. Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLoS Genet 11: e1004901.
[160] Shimizu, T., H. Marusawa, Y. Endo, and T. Chiba. 2012. Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci 103: 1201–1206.
[161] Karin, M., and F. R. Greten. 2005. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5: 749–759.
[162] Grivennikov, S. I., F. R. Greten, and M. Karin. 2010. Immunity, inflammation, and cancer. Cell 140: 883–899.
[163] You, Z., L. V. Madrid, D. Saims, J. Sedivy, and C. Y. Wang. 2002. c-Myc sensitizes cells to tumor necrosis factor-mediated apoptosis by inhibiting nuclear factor kappa B transactivation. J Biol Chem 277: 36671–36677.
[164] Pires, B. R., A. L. Mencalha, G. M. Ferreira, W. F. de Souza, J. A. Morgado-Díaz, A. M. Maia, S. Corrêa, and E. S. Abdelhay. 2017. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS One 12: e0169622.
[165] Wu, Y., and B. P. Zhou. 2009. Inflammation: a driving force speeds cancer metastasis. Cell Cycle 8: 3267–3273.
[166] Gilkes, D. M., and G. L. Semenza. 2013. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol 9: 1623–1636.
[167] Spina, A., V. De Pasquale, G. Cerulo, P. Cocchiaro, R. Della Morte, L. Avallone, and L. M. Pavone. 2015. HGF/c-MET Axis in Tumor Microenvironment and Metastasis Formation. Biomedicines 3: 71–88.
[168] Balkwill, F. 2004. Cancer and the chemokine network. Nat Rev Cancer 4: 540–550.
[169] Vereecke, L., R. Beyaert, and G. van Loo. 2009. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30: 383–391.
[170] Heyninck, K., D. De Valck, W. Vanden Berghe, W. Van Criekinge, R. Contreras, W. Fiers, G. Haegeman, and R. Beyaert. 1999. The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol 145: 1471–1482.
[171] Nakagawa, M. M., K. Thummar, J. Mandelbaum, L. Pasqualucci, and C. V. Rathinam. 2015. Lack of the ubiquitin-editing enzyme A20 results in loss of hematopoietic stem cell quiescence. J Exp Med 212: 203–216.
[172] Sitcheran, R., P. C. Cogswell, and A. S. Baldwin, Jr. 2003. NF-kappaB mediates inhibition of mesenchymal cell differentiation through a posttranscriptional gene silencing mechanism. Genes Dev 17: 2368–2373.
[173] Guttridge, D. C., M. W. Mayo, L. V. Madrid, C. Y. Wang, and A. S. Baldwin, Jr. 2000. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289: 2363–2366.
[174] Kaltschmidt, C., J. F. W. Greiner, and B. Kaltschmidt. 2021. The Transcription Factor NF-B in Stem Cells and Development. Cells 10: 2042.
[175] Chan, Y. H., Y. C. Lee, C. Y. Hung, P. J. Yang, P. C. Lai, and S. W. Feng. 2021. Three-dimensional Spheroid Culture Enhances Multipotent Differentiation and Stemness Capacities of Human Dental Pulp-derived Mesenchymal Stem Cells by Modulating MAPK and NF-kB Signaling Pathways. Stem Cell Rev Rep 17: 1810–1826.
[176] Proto, J. D., A. Lu, A. Dorronsoro, A. Scibetta, P. D. Robbins, L. J. Niedernhofer, and J. Huard. 2017. Inhibition of NF-B improves the stress resistance and myogenic differentiation of MDSPCs isolated from naturally aged mice. PLoS One 12: e0179270.
[177] Greco, S. J., S. V. Smirnov, R. G. Murthy, and P. Rameshwar. 2007. Synergy between the RE-1 silencer of transcription and NFkappaB in the repression of the neurotransmitter gene TAC1 in human mesenchymal stem cells. J Biol Chem 282: 30039–30050.
[178] Buhrmann, C., A. Mobasheri, U. Matis, and M. Shakibaei. 2010. Curcumin mediated suppression of nuclear factor-B promotes chondrogenic differentiation of mesenchymal stem cells in a high-density co-culture microenvironment. Arthritis Res Ther 12: R127.
[179] Langen, R. C., A. M. Schols, M. C. Kelders, E. F. Wouters, and Y. M. Janssen-Heininger. 2001. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-kappaB. FASEB J 15: 1169–1180.
[180] Hess, K., A. Ushmorov, J. Fiedler, R. E. Brenner, and T. Wirth. 2009. TNFalpha promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-kappaB signaling pathway. Bone 45: 367–376.
[181] Lu, Z., G. Wang, C. R. Dunstan, and H. Zreiqat. 2012. Short-term exposure to tumor necrosis factor-alpha enables human osteoblasts to direct adipose tissue-derived mesenchymal stem cells into osteogenic differentiation. Stem Cells Dev 21: 2420–2429.
[182] Lombardo, E., O. DelaRosa, P. Mancheño-Corvo, R. Menta, C. Ramírez, and D. Büscher. 2009. Toll-like receptor-mediated signaling in human adipose-derived stem cells: implications for immunogenicity and immunosuppressive potential. Tissue Eng Part A 15: 1579–1589.
[183] Vazquez-Santillan, K., J. Melendez-Zajgla, L. Jimenez-Hernandez, G. Martínez-Ruiz, and V. Maldonado. 2015. NF-B signaling in cancer stem cells: a promising therapeutic target? Cell Oncol (Dordr) 38: 327–339.
[184] Shigdar, S., Y. Li, S. Bhattacharya, M. O’Connor, C. Pu, J. Lin, T. Wang, D. Xiang, L. Kong, M. Q. Wei, Y. Zhu, S. Zhou, and W. Duan. 2014. Inflammation and cancer stem cells. Cancer Lett 345: 271–278.
[185] Rinkenbaugh, A. L., and A. S. Baldwin. 2016. The NF-B Pathway and Cancer Stem Cells. Cells 5.
[186] Wendt, M. K., N. Balanis, C. R. Carlin, and W. P. Schiemann. 2014. STAT3 and epithelial-mesenchymal transitions in carcinomas. Jakstat 3: e28975.
[187] Yamamoto, M., Y. Taguchi, T. Ito-Kureha, K. Semba, N. Yamaguchi, and J. Inoue. 2013. NF-B non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun 4: 2299.
[188] Guzman, M. L., S. J. Neering, D. Upchurch, B. Grimes, D. S. Howard, D. A. Rizzieri, S. M. Luger, and C. T. Jordan. 2001. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98: 2301–2307.
[189] Mitsiades, C. S., N. Mitsiades, V. Poulaki, R. Schlossman, M. Akiyama, D. Chauhan, T. Hideshima, S. P. Treon, N. C. Munshi, P. G. Richardson, and K. C. Anderson. 2002. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene 21: 5673–5683.
[190] Zhou, J., J. Y. Chooi, Y. Q. Ching, J. Y. Quah, S. H. Toh, Y. Ng, T. Z. Tan, and W. J. Chng. 2018. NF-B promotes the stem-like properties of leukemia cells by activation of LIN28B. World J Stem Cells 10: 34–42.
[191] Gallipoli, P., F. Pellicano, H. Morrison, K. Laidlaw, E. K. Allan, R. Bhatia, M. Copland, H. G. Jørgensen, and T. L. Holyoake. 2013. Autocrine TNF- production supports CML stem and progenitor cell survival and enhances their proliferation. Blood 122: 3335–3339.
[192] Chefetz, I., A. B. Alvero, J. C. Holmberg, N. Lebowitz, V. Craveiro, Y. Yang-Hartwich, G. Yin, L. Squillace, M. Gurrea Soteras, P. Aldo, and G. Mor. 2013. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle 12: 511–521.
[193] Burnstock, G. 2018. Purine and purinergic receptors. Brain Neurosci Adv 2: 2398212818817494.
[194] Haskó, G., and B. Cronstein. 2013. Regulation of inflammation by adenosine. Front Immunol 4: 85.
[195] North, R. A. 2002. Molecular physiology of P2X receptors. Physiol Rev 82: 1013–1067.
[196] Burnstock, G., and G. E. Knight. 2004. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240: 31–304.
[197] Burnstock, G. 2006. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev 58: 58–86.
[198] Verkhratsky, A., and G. Burnstock. 2014. Biology of purinergic signalling: its ancient evolutionary roots, its omnipresence and its multiple functional significance. Bioessays 36: 697–705.
[199] Vadlamani, V. M. K., K. K. J. Gunasinghe, X. W. Chee, T. Rahman, and M. T. Harper. 2023. Human soluble CD39 displays substrate inhibition in a substrate-specific manner. Sci Rep 13: 8958.
[200] Giuliani, A. L., A. C. Sarti, and F. Di Virgilio. 2020. Ectonucleotidases in Acute and Chronic Inflammation. Front Pharmacol 11: 619458.
[201] Allard, B., M. S. Longhi, S. C. Robson, and J. Stagg. 2017. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev 276: 121–144.
[202] Burnstock, G., and C. Kennedy. 1985. Is there a basis for distinguishing two types of P2-purinoceptor? Gen Pharmacol 16: 433–440.
[203] Fredholm, B. B., I. J. AP, K. A. Jacobson, K. N. Klotz, and J. Linden. 2001. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53: 527–552.
[204] Abbracchio, M. P., and G. Burnstock. 1994. Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol Ther 64: 445–475.
[205] Huang, Z., N. Xie, P. Illes, F. Di Virgilio, H. Ulrich, A. Semyanov, A. Verkhratsky, B. Sperlagh, S. G. Yu, C. Huang, and Y. Tang. 2021. From purines to purinergic signalling: molecular functions and human diseases. Signal Transduct Target Ther 6: 162.
[206] Duarte-Silva, E., H. Ulrich, Á. Oliveira-Giacomelli, H. P. Hartung, S. G. Meuth, and C. A. Peixoto. 2022. The adenosinergic signaling in the pathogenesis and treatment of multiple sclerosis. Front Immunol 13: 946698.
[207] Passarella, D., M. Ronci, V. Di Liberto, M. Zuccarini, G. Mudò, C. Porcile, M. Frinchi, P. Di Iorio, H. Ulrich, and C. Russo. 2022. Bidirectional Control between Cholesterol Shuttle and Purine Signal at the Central Nervous System. Int J Mol Sci 23.
[208] Tomczyk, M., T. Glaser, E. M. Slominska, H. Ulrich, and R. T. Smolenski. 2021. Purine Nucleotides Metabolism and Signaling in Huntington’s Disease: Search for a Target for Novel Therapies. Int J Mol Sci 22: 6545.
[209] Li, Y., X. Han, J. Tong, Y. Wang, X. Liu, Z. Liao, M. Jiang, and H. Zhao. 2023. Analysis of Metabolites in Gout: A Systematic Review and Meta-Analysis. Nutrients 15: 3143.
[210] Gilbert, S. M., C. J. Oliphant, S. Hassan, A. L. Peille, P. Bronsert, S. Falzoni, F. Di Virgilio, S. McNulty, and R. Lara. 2019. ATP in the tumour microenvironment drives expression of nfP2X(7), a key mediator of cancer cell survival. Oncogene 38: 194–208.
[211] Burnstock, G., and F. Di Virgilio. 2013. Purinergic signalling and cancer. Purinergic Signal 9: 491–540.
[212] Jia, W., Z. Huang, L. Zhou, Y. C. Liou, F. Di Virgilio, H. Ulrich, P. Illes, W. Zhang, C. Huang, and Y. Tang. 2023. Purinergic signalling in cancer therapeutic resistance: From mechanisms to targeting strategies. Drug Resist Updat 70: 100988.
[213] He, J. R., S. G. Yu, Y. Tang, and P. Illes. 2020. Purinergic signaling as a basis of acupuncture-induced analgesia. Purinergic Signal 16: 297–304.
[214] Deaglio, S., K. M. Dwyer, W. Gao, D. Friedman, A. Usheva, A. Erat, J. F. Chen, K. Enjyoji, J. Linden, M. Oukka, V. K. Kuchroo, T. B. Strom, and S. C. Robson. 2007. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204: 1257–1265.
[215] Linden, J., F. Koch-Nolte, and G. Dahl. 2019. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu Rev Immunol 37: 325–347.
[216] Bissacotti, B. F., P. M. Copetti, N. B. Bottari, T. V. Palma, M. M. Pillat, C. M. de Andrade, V. M. M. Morsch, H. Ulrich, and A. S. da Silva. 2023. Curcumin modulates neurogliogenesis and purinergic receptor expression in neural precursor cells infected with Toxoplasma gondii. Parasitol Res 122: 77–84.
[217] Franczak, S., H. Ulrich, and M. Z. Ratajczak. 2025. Hematopoietic stem cells on the crossroad between purinergic signaling and innate immunity. Purinergic Signal 21: 3–9.
[218] Ratajczak, M. Z., M. Adamiak, K. Bujko, A. Thapa, V. Pensato, M. Kucia, J. Ratajczak, and H. Ulrich. 2020. Innate immunity orchestrates the mobilization and homing of hematopoietic stem/progenitor cells by engaging purinergic signaling-an update. Purinergic Signal 16: 153–166.
[219] Sheth, S., R. Brito, D. Mukherjea, L. P. Rybak, and V. Ramkumar. 2014. Adenosine receptors: expression, function and regulation. Int J Mol Sci 15: 2024–2052.
[220] Bertolini, G., M. Compagno, D. C. Belisario, C. Bracci, T. Genova, F. Mussano, M. Vitale, A. Horenstein, F. Malavasi, R. Ferracini, and I. Roato. 2022. CD73/Adenosine Pathway Involvement in the Interaction of Non-Small Cell Lung Cancer Stem Cells and Bone Cells in the Pre-Metastatic Niche. Int J Mol Sci 23.
[221] Liu, R., W. Chang, H. Wei, and K. Zhang. 2016. Comparison of the Biological Characteristics of Mesenchymal Stem Cells Derived from Bone Marrow and Skin. Stem Cells Int 2016: 3658798.
[222] Torres, A., Y. Vargas, D. Uribe, C. Jaramillo, A. Gleisner, F. Salazar-Onfray, M. N. López, R. Melo, C. Oyarzún, R. San Martín, and C. Quezada. 2016. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 7: 67373–67386.
[223] Uhlén, M., L. Fagerberg, B. M. Hallström, C. Lindskog, P. Oksvold, A. Mardinoglu, Å. Sivertsson, C. Kampf, E. Sjöstedt, A. Asplund, I. Olsson, K. Edlund, E. Lundberg, S. Navani, C. A. Szigyarto, J. Odeberg, D. Djureinovic, J. O. Takanen, S. Hober, T. Alm, P. H. Edqvist, H. Berling, H. Tegel, J. Mulder, J. Rockberg, P. Nilsson, J. M. Schwenk, M. Hamsten, K. von Feilitzen, M. Forsberg, L. Persson, F. Johansson, M. Zwahlen, G. von Heijne, J. Nielsen, and F. Pontén. 2015. Proteomics. Tissue-based map of the human proteome. Science 347: 1260419.
[224] Sattler, C., M. Steinsdoerfer, M. Offers, E. Fischer, R. Schierl, K. Heseler, W. Däubener, and J. Seissler. 2011. Inhibition of T-cell proliferation by murine multipotent mesenchymal stromal cells is mediated by CD39 expression and adenosine generation. Cell Transplant 20: 1221–1230.
[225] Eltzschig, H. K., J. C. Ibla, G. T. Furuta, M. O. Leonard, K. A. Jacobson, K. Enjyoji, S. C. Robson, and S. P. Colgan. 2003. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med 198: 783–796.
[226] Kaebisch, C., D. Schipper, P. Babczyk, and E. Tobiasch. 2015. The role of purinergic receptors in stem cell differentiation. Comput Struct Biotechnol J 13: 75–84.
[227] Carroll, S. H., N. A. Wigner, N. Kulkarni, H. Johnston-Cox, L. C. Gerstenfeld, and K. Ravid. 2012. A2B adenosine receptor promotes mesenchymal stem cell differentiation to osteoblasts and bone formation in vivo. J Biol Chem 287: 15718–15727.
[228] He, W., A. Mazumder, T. Wilder, and B. N. Cronstein. 2013. Adenosine regulates bone metabolism via A1, A2A, and A2B receptors in bone marrow cells from normal humans and patients with multiple myeloma. FASEB J 27: 3446–3454.
[229] Shih, Y. R., Y. Hwang, A. Phadke, H. Kang, N. S. Hwang, E. J. Caro, S. Nguyen, M. Siu, E. A. Theodorakis, N. C. Gianneschi, K. S. Vecchio, S. Chien, O. K. Lee, and S. Varghese. 2014. Calcium phosphate-bearing matrices induce osteogenic differentiation of stem cells through adenosine signaling. Proc Natl Acad Sci U S A 111: 990–995.
[230] Gharibi, B., A. A. Abraham, J. Ham, and B. A. Evans. 2012. Contrasting effects of A1 and A2b adenosine receptors on adipogenesis. Int J Obes (Lond) 36: 397–406.
[231] Soliman, H., M. Theret, W. Scott, L. Hill, T. M. Underhill, B. Hinz, and F. M. V. Rossi. 2021. Multipotent stromal cells: One name, multiple identities. Cell Stem Cell 28: 1690–1707.
[232] Patel, N., T. E. Klassert, S. J. Greco, S. A. Patel, J. L. Munoz, B. Y. Reddy, M. Bryan, N. Campbell, N. Kokorina, H. E. Sabaawy, and P. Rameshwar. 2012. Developmental regulation of TAC1 in peptidergic-induced human mesenchymal stem cells: implication for spinal cord injury in zebrafish. Stem Cells Dev 21: 308–320.
[233] da Silva Meirelles, L., A. I. Caplan, and N. B. Nardi. 2008. In search of the in vivo identity of mesenchymal stem cells. Stem Cells 26: 2287–2299.
[234] Kusiak, A., and G. Brady. 2022. Bifurcation of signalling in human innate immune pathways to NF-kB and IRF family activation. Biochem Pharmacol 205: 115246.
[235] Zinatizadeh, M. R., B. Schock, G. M. Chalbatani, P. K. Zarandi, S. A. Jalali, and S. R. Miri. 2021. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis 8: 287–297.
[236] Li, Y., X. Wang, and J. Lu. 2023. Interleukin-35 Promote Osteogenesis and Inhibit Adipogenesis: Role of Wnt/-Catenin and PPAR Signaling Pathways. Inflammation 46: 522–533.
[237] Das, R., J. Giri, K. P. P, N. Froelich, R. Chinnadurai, S. McCoy, W. Bushman, and J. Galipeau. 2022. A STAT5-Smad3 dyad regulates adipogenic plasticity of visceral adipose mesenchymal stromal cells during chronic inflammation. Regen Med 7: 41.
[238] Zhang, Y., L. Ling, D. O. A. A. Ajay, Y. M. Eio, A. J. van Wijnen, V. Nurcombe, and S. M. Cool. 2022. FGFR2 accommodates osteogenic cell fate determination in human mesenchymal stem cells. Gene 818: 146199.
[239] Yoon, D. S., Y. H. Kim, S. Lee, K. M. Lee, K. H. Park, Y. Jang, and J. W. Lee. 2014. Interleukin-6 induces the lineage commitment of bone marrow-derived mesenchymal multipotent cells through down-regulation of Sox2 by osteogenic transcription factors. FASEB J 28: 3273–3286.
[240] Iwata, T., N. Mizuno, T. Nagahara, E. Kaneda-Ikeda, M. Kajiya, S. Sasaki, K. Takeda, M. Kiyota, R. Yagi, T. Fujita, and H. Kurihara. 2022. Cytokines regulate stemness of mesenchymal stem cells via miR-628–5p during periodontal regeneration. J Periodontol 93: 269–286.
[241] Gu, Y., X. Ding, J. Huang, M. Xue, J. Zhang, Q. Wang, H. Yu, Y. Wang, F. Zhao, H. Wang, M. Jin, Y. Wu, and Y. Zhang. 2018. The deubiquitinating enzyme UCHL1 negatively regulates the immunosuppressive capacity and survival of multipotent mesenchymal stromal cells. Cell Death Dis 9: 459.
[242] Yuan, X., D. Ferrari, T. Mills, Y. Wang, A. Czopik, M. F. Doursout, S. E. Evans, M. Idzko, and H. K. Eltzschig. 2021. Editorial: Purinergic Signaling and Inflammation. Front Immunol 12: 699069.
[243] Rossi, L., V. Salvestrini, D. Ferrari, F. Di Virgilio, and R. M. Lemoli. 2012. The sixth sense: hematopoietic stem cells detect danger through purinergic signaling. Blood 120: 2365–2375.
[244] Scarfì, S. 2014. Purinergic receptors and nucleotide processing ectoenzymes: Their roles in regulating mesenchymal stem cell functions. World J Stem Cells 6: 153–162.
[245] Sun, Y., W. Hu, X. Yu, Z. Liu, R. Tarran, K. Ravid, and P. Huang. 2016. Actinin-1 binds to the C-terminus of A2B adenosine receptor (A2BAR) and enhances A2BAR cell-surface expression. Biochem J 473: 2179–2186.
[246] Zhao, N., G. Xia, J. Cai, Z. Li, and X. W. Lv. 2022. Adenosine receptor A2B mediates alcoholic hepatitis by regulating cAMP levels and the NF-KB pathway. Toxicol Lett 359: 84–95.
[247] Murphy, P. S., J. Wang, S. P. Bhagwat, J. C. Munger, W. J. Janssen, T. W. Wright, and M. R. Elliott. 2017. CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. Cell Death Differ 24: 559–570.
[248] Bauer, F. N., T. Tertel, O. Stambouli, C. Wang, R. Dittrich, S. Staubach, V. Börger, D. M. Hermann, S. Brandau, and B. Giebel. 2023. CD73 activity of mesenchymal stromal cell-derived extracellular vesicle preparations is detergent-resistant and does not correlate with immunomodulatory capabilities. Cytotherapy 25: 138–147.
[249] Burand, A. J., Jr., L. Di, L. K. Boland, D. T. Boyt, M. V. Schrodt, D. A. Santillan, and J. A. Ankrum. 2020. Aggregation of Human Mesenchymal Stromal Cells Eliminates Their Ability to Suppress Human T Cells. Front Immunol 11: 143.
[250] Grünewald, J. K., and A. J. Ridley. 2010. CD73 represses pro-inflammatory responses in human endothelial cells. J Inflamm (Lond) 7: 10.
[251] Tan, K., H. Zhu, J. Zhang, W. Ouyang, J. Tang, Y. Zhang, L. Qiu, X. Liu, Z. Ding, and X. Deng. 2019. CD73 Expression on Mesenchymal Stem Cells Dictates the Reparative Properties via Its Anti-Inflammatory Activity. Stem Cells Int 2019: 8717694.
[252] Yang, J., X. Liao, J. Yu, and P. Zhou. 2018. Role of CD73 in Disease: Promising Prognostic Indicator and Therapeutic Target. Curr Med Chem 25: 2260–2271.
[253] Ramos, T. L., L. I. Sánchez-Abarca, B. Rosón-Burgo, A. Redondo, A. Rico, S. Preciado, R. Ortega, C. Rodríguez, S. Muntión, Á. Hernández-Hernández, J. De Las Rivas, M. González, J. R. González Porras, C. Del Cañizo, and F. Sánchez-Guijo. 2017. Mesenchymal stromal cells (MSC) from JAK2+ myeloproliferative neoplasms differ from normal MSC and contribute to the maintenance of neoplastic hematopoiesis. PLoS One 12: e0182470.
International Journal of Translational Science, Vol. 1, 25–68.
doi: 10.13052/ijts2246-8765.2025.003
© 2025 River Publishers