Intercellular Interaction Between a Dental Pulp Stem Cell and Leukemia Cells – Support of Acute Myeloid Leukemia
Ioanna Tsolaki1, 2,*, Lauren S. Sherman2, Darling P. Rojas1, 2, Yahaira Naaldijk2, Andrew Petryna1, Yannick Kenfack2, Garima Sinha2, Vincent Tsiagbe3 and Pranela Rameshwar2,*
1Rutgers School of Dental Medicine, Department of Periodontics, Newark, NJ, United States
2Rutgers New Jersey Medical School, Department of Medicine, Newark, NJ, United States
3Rutgers School of Dental Medicine, Department of Oral Biology, Newark, NJ, United States
E-mail: it120@sdm.rutgers.edu; rameshwa@njms.rutgers.edu
*Corresponding Authors
Received 02 May 2025; Accepted 03 June 2025
Abstract
The oral cavity is a site of hematopoietic activity, and metastatic hematological and solid tumors. This study focused on acute myeloid leukemia (AML) due to extensive documentation of its presentation in the gingiva. Despite these reports, it is unclear how AML and other leukemia cells survive in the oral tissue. We investigated intercellular communication between leukemia cells and dental pulp stem cells (DPSCs). DPSCs enhanced the proliferation and adhesion of AML cells (HL-60) and to a lesser extent, myelomoncytic leukemia cells (U937). The erythroleukemia K562 cells showed a delayed trend to proliferate. The communication between DPSCs and HL-60 cells was partly due to gap junctional intercellular communication (GJIC), as indicated by dye transfer. We also noted evidence of tunnelling nanotubules (TNT). Dye transfer was noted in non-adherent cells, suggesting other method of transfer, perhaps by extracellular vesicles. Using SORE-6 that can stratify the HL-60 subset, dye transfer occurred mostly in the subset lowest in the hierarchy. These latter findings were novel since they might provide insights into the behavior of non-leukemia stem cells and their interaction with cells in the oral cavity. In summary, this study began to dissect the interaction between HL-60 AML cells and DPSCs, providing insights into the survival of AML and perhaps other leukemia cells in the oral cavity.
Keywords: Leukemia, stem cell, dental pulp, gap junction.
Introduction
The oral cavity is a metastatic site for hematological and solid tumors [1]. At least 20 solid tumors have been reported to metastasize in the soft and hard tissue of the oral cavity [2]. Similarly, hematological tumors – leukemia, lymphoma, and multiple myeloma – have been reported in the oral cavity [3].
The oral cavity has differentiated specialized cell types, and stem cells. Of note is the center of a tooth with a well vascularized region, referred to as the dental pulp. The pulp is present within the crown and the root of the tooth. It serves functions related to sensation, nourishment, and tissue regeneration with the major nerves and microvasculature to nourish the tooth [4]. The neurovascular bundle enters and exits the tooth structure through an apical foramen at the root of the tooth. The superior and inferior alveolar arteries are responsible for perfusing the maxilla and mandible, including associated structures like the teeth and the gingiva, illustrating the closely interconnected relationship between pulpal and gingival circulation [4].
The dental pulp consists of loose connective tissue that contains fibrous matrix (predominantly collagen fibers type I and II), ground substance (proteoglycans, glycoproteins, and water), and various cell types including stem cells (DPSCs), fibroblasts, endothelial cells, odontoblasts, Schwann cells, differentiated hematological cells such as immune and erythrocytes, and epithelial-like cells. Vascularization of the dental pulp is critical, with healthy pulp of different teeth showing similar ranges of oxygen saturation [5].
Acute myeloid leukemia (AML) is one of several hematological malignancies that can survive in the oral cavity. Despite improved treatment and diagnosis, the prognosis of AML is poor with a five-year survival rate of about 46% [6]. AML is presented in the gingival tissue with overgrowth and potential ulceration [7–13]. Such presentation is similar to thrombocytopenia and immunodeficiency [14]. All subtypes of AML can present with oral manifestations which include various degrees of gingival overgrowth, petechiae, spontaneous bleeding, and mucosal ulceration, with or without necrosis [15, 16]. AML gingival overgrowth is commonly seen with the FAB AML subtypes of acute monocytic leukemia (M5) and acute myelomonocytic leukemia (M4) [17–26]. Interestingly, gingival infiltration is the first clinical sign of AML in 5% of AML patients [15, 19, 27, 28]. The microanatomy, combined with the expression of endothelial adhesion molecules, contributed to the infiltration of malignant and non-malignant hematopoietic cells [15]. This results in an increase of the gingival volume, leading to periodontal pockets and pseudo- pockets. The interaction between leukemic gingival infiltration and periodontal diseases, i.e. gingivitis and periodontitis, enhances the symptoms of each other. Despite the extensive documentation of the clinical presentation of AML in the gingiva and the histologic confirmation of the gingival AML infiltration, there is a gap in understanding the mechanisms by which the oral tissue supports AML. This critical gap in knowledge could impact early diagnosis and treatment for prolonged remission of AML subjects. This gap is partly due to the current and past emphasis in addressing the clinical and research landscape of AML pathology in the bone marrow (BM), blood and lymphatic.
Currently, it is unclear if current treatment for AML can be applied to the oral cavity. Insights were gained from clinical cases in which AML relapse showed cancer cells within the gingiva but undetectable blast in the BM [29, 30]. This suggested that AML could relapse from the gingiva, in addition to the BM. Another case report showed gingival overgrowth of AML two years after remission with negative results in the BM biopsy [30]. Taken together, these case reports led the inevitable question, does the oral tissue have the potential to serve as a site of dormancy for the AML or metastatic site? If so, how are AML cells supported in the oral tissue? Furthermore, are current treatments effective for AML in the oral cavity?
Despite several reports on AML infiltrating the gingiva, it is unclear if these tumors can enter the dental pulp. The literature has indicated a supporting role for DPSCs in solid tumors [31–34]. The dental pulp niche presents with interesting similarities to the BM with areas of hypoxia, abundance of endothelial cells and endothelial progenitors, MSCs, and hematopoietic cells [35]. The anatomic structure of dental pulp parallels that of the BM with respect to cell types and vascularity. This has led to the question of whether DPSCs function as a major cell support of AML in the oral cavity (Supplemental Figure 1).
This study is relevant and impactful since the dentist, oftentimes, is the first healthcare provider to notice the gingival overgrowth for referral to the hematologist. The dentist is also responsible for treating the cancer patient both periodontally and restoratively prior to any planned chemotherapy or radiotherapy. Currently, there is no information on the response of AML in the oral cavity to current treatment. Since AML aggressively infiltrates the surrounding gingival tissue, most probably it also affects the dental pulp, which is experimentally addressed in this study.
Materials and Methods
Reagents
RPMI 1640, glutamine, penicillin-streptomycin, fungizone, 1-octanol, fetal calf sera (FCS) and phosphate buffered saline (PBS) were purchased from Millipore-Sigma (St Louis, MO). Cell Tracker Deep Red, polybrene, StemPro adipogenesis and osteogenic differentiation kits were purchased from Thermo Fisher Scientific (Waltham, MA). The following fluorescein conjugated antibodies and corresponding isotype control were purchased from BD Bioscience (San Jose, CA): murine anti-human – CD105-PE, CD44-PE, -CD45-FITC and -CD90-APC. Murine anti-human Ki67 and anti-murine IgG-PE were obtained from Abcam (Waltman, MA).
Ethics Statement
Rutgers Institutional Review Board (IRB) approved the use of dental pulp. The demographics of the donors are shown in Table 1.
Table 1 Demographics of teeth from healthy and periodontal disease donors
| Tooth | Periodontal | |||||
| Patient | Tooth # | Age | Gender | Race | Vitality | Status |
| 1 | 5, 12, 21, 28 | 17 | M | African American | Vital | Healthy |
| 2 | 5, 12, 21, 28 | 14 | F | Asian | Vital | Healthy |
| 3 | 28 | 21 | F | Hispanic | Vital | Healthy |
| 4 | 5, 6 | 49 | M | Caucasian | Vital | Periodontitis |
| 5 | 5, 12 | 33 | M | African American | Vital | Gingivitis |
| 6 | 6, 8 | 66 | M | Caucasian | Vital | Healthy |
| 7 | 5, 12 | 18 | F | African American | Vital | Gingivitis |
| 8 | 21 | 18 | F | Hispanic | Vital | Healthy |
Cell Lines
The following cell lines were obtained from American Type Culture Collection (ATCC) (Manassas, VA): HL-60 AML cells, myelomonocytic U937 cells, erythroleukemia K562 cells and HEK-293-T. The cells were cultured as per ATCC instruction. K562, HL-60 and U937 were cultured in RPMI 1640 with 10% FCS.
Vector
The source and preparation of the lentiviral plasmids for SORE6-GFP has previously been described [36]. SORE6-GFP was propagated in HEK-293-T cells. HL-60 cells were stably transfected with SORE6-GFP similar to the described method for the glioblastoma cell line. Briefly, HL-60 were transduced with SORE6-GFP or empty vector-GFP lentivirus at multiplicity of infection (MOI) of 5:1 with 0.8 g/l of polybrene. Stable transductions were selected with 1–3 g/l of puromycin. The stable cell lines were maintained with 1 g/l puromycin for SORE6-GFP, and 2 g/l puromycin for cells with empty vector.
Culture of DPSCs
DPSCs were cultured from freshly extracted teeth. The extracted tooth was stored in sterile 1X PBS for transit from the clinic to the laboratory and rinsed with 1X PBS prior to processing. A high-speed dental hand-piece with a sterile carbide bur was used to drill grooves into the dentin along the buccal and lingual/palatal surface of each of the roots and the crown of the tooth, as well as at the perimeter of the anatomic crown of the tooth at the level of the cemento–enamel junction. The prepared tooth was then decontaminated by serially incubating in pure Povidone-iodine for 4 min; sterile 1X PBS containing 100 IU/ml penicillin and 100 g/ml streptomycin for 4 min, exchanging the solution until the iodine was removed; Fungizone (0.25 g/ml) for 4 min; and finally rinsed three times with 1X PBS.
The disinfected tooth was fractured along the prepared grooves using a dental mallet and chisel. The dental pulp was extracted using Micro Adson tissue pliers and periodontal probe NC15, minced into pieces of 1–5 mm in length, evenly distributed throughout a vacuum gas plasma-treated 60 mm petri dish, and allowed to dry for 10 min. The dried pulp was covered with DMEM containing 10% FBS, 100 IU/ml penicillin, 100 g/ml streptomycin, and 2 mM L-glutamine and incubated at 37C with 5% CO. The media were replaced with 50% fresh media at 3–4 day intervals until the cells reached 70–80% confluency. At passage 3, the cells were characterized for phenotype and multipotency, as described [37].
DPSC Differentiation
DPSCs were differentiated into adipogenic and osteogenic lineages using the listed kits. Induction was performed based on manufactures’ protocol. Briefly DPSCs/cm were transferred to Falcon tissue cultured vacuum gas plates and allowed to adhere. Cells were plated in 10% DMEM media for 24 h. DMEM was replaced by induction media and incubated for 72 h at 37C in a CO incubator. Cells were washed followed by adding of maintenance media. After adipogenesis induction, the cells were incubated with 0.18% oil red solution. Red stain cellular uptake was observed using bright-field microscopy at 10 magnification.
Flow Cytometry
DPSCs were labeled with anti-CD44-PE, -CD45-FITC and -CD90-APC, each at 1/100 dilution. The DPSCs were incubated in the cold for 30 mins, followed by washing in PBS twice. The cells were resuspended in 1% formaldehyde and then immediately acquired on the FACScan (BD Bioscience). The data were analyzed with Flowjo (Flowjo, LLC; San Carlos, CA).
Dye Transfer Assay
GJIC between DPSCs and leukemic cells or BC cells were performed as described [1]. DPSCs were labeled with 2.5 mM of CellTracker Deep Red dye. This was accomplished by incubating the cells with the dye for 30 mins at 37C in a CO incubator. After incubation, the cells were washed twice with 1X PBS. Co-cultures of dye-loaded DPSCs and cancer cells were at 1:1 ratio. After 24 h, the cancer cells were assessed for GJIC by flow cytometry. Control co-cultures used 1-octanol (300 M). Dye transfer by flow cytometry occurred after 72 h as follows: cells were trypsinized, pelleted by centrifugation, and then resuspended in 0.5 mL 1X PBS.
Western Blot
Cells were resuspended in 50–100 l of lysis buffer and EDTA-protease inhibitor cocktail (Millipore-Sigma). Cell lysates were subjected to freeze/thaw cycles – 2 mins in liquid nitrogen followed by 2 mins in a 37C water bath. Cell lysates were centrifuged at 2000 g for 10 mins and the supernatants containing the proteins were collected and then quantitated for protein levels with the Bradford Protein Assay Reagent (BioRad, Hercules, CA). Extracts (15 mg) were electrophoresed on 12% SDS-PAGE gel and then transferred onto Immobilon-P PVDF membranes (ThermoFisher Scientific). The membranes were washed with 1X PBS-tween for 10 mins and then blocked for 20 mins with 3% non-fat milk diluted in 1X PBS. The membranes were incubated overnight at 4C on a shaker with the primary antibodies – anti-Cx43 at a 1:1000 ratio in 3% non-fat milk. Membranes were washed and blocked with 3% non-fat milk, followed by incubation with HRP tagged anti--actin at 1:2000 in 3% non-fat milk for 2 h at 4C. The membranes were washed for 20 mins and then developed with the Super Signal West Femto Maximum Sensitivity Substrate for 5 mins. The protein bands were imaged using the using the ChemiDoc XRA (BioRad).
Co-culture of DPSCs and Tumor Cells (Adhesion, Proliferation)
DPSCs, , were added to six-well tissue culture plates. After overnight incubation, equal numbers of leukemia cells were added. After 24, 48, and 72 h, the cells were labeled for Ki67 at 0.5 g/mL to assess proliferating cells. The cultures were imaged on EVOS FL and Ki67 cells counted for proliferation. The adherent cells were assessed after removing the non-adherent cells and washing the wells with PBS.
Statistical Analysis
Data was analyzed using Student’s t-test and ANOVA to compare between groups. A p value of 0.05 was considered significant.
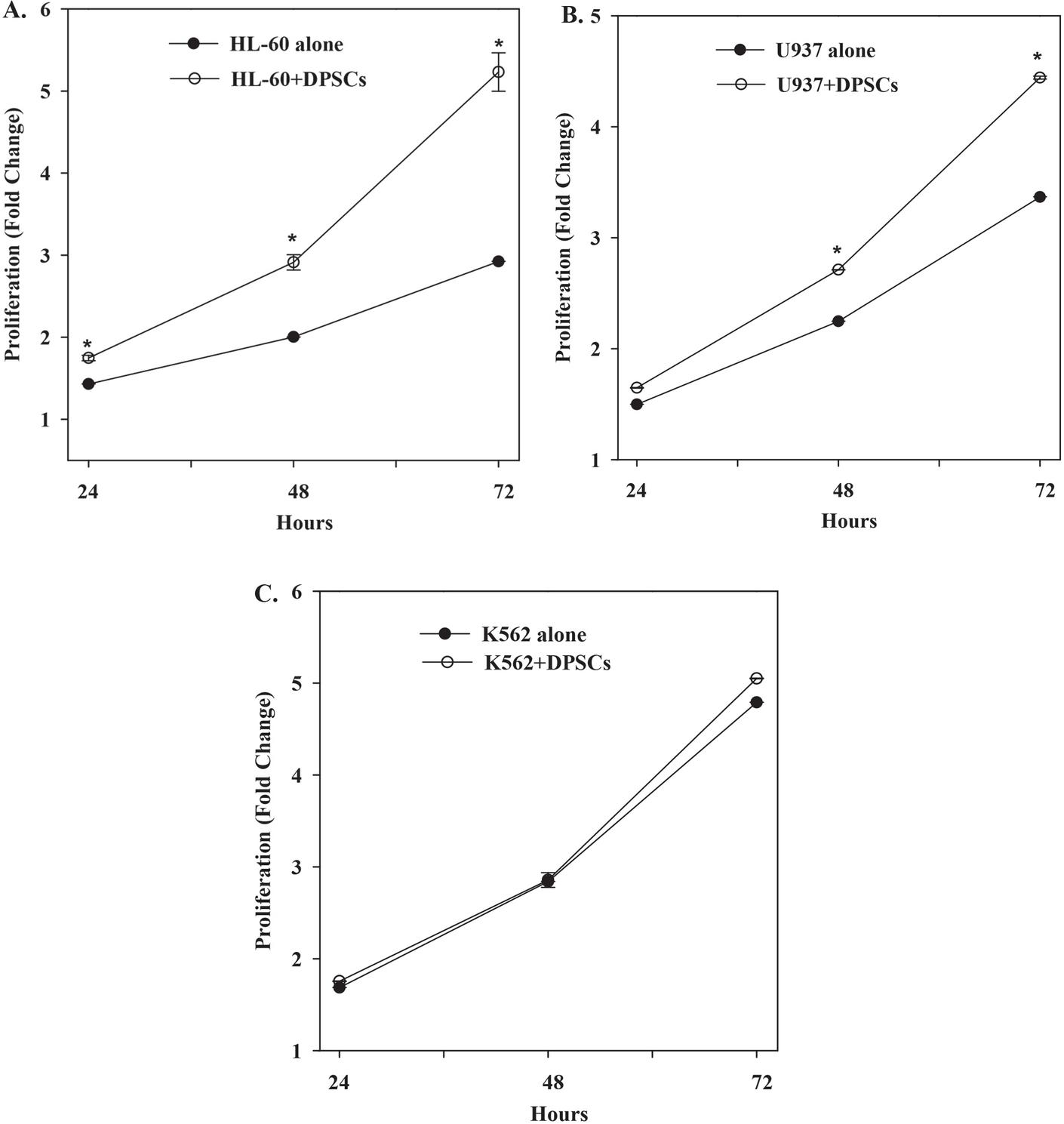
Figure 1 Timeline proliferation of leukemic cells in the presence or absence of DPSCs. The results are shown for three biological replicates SD. (A) HL-60 cells were seeded at equal ratio with DPSCs. The total number of HL-60 cells were counted at different times. (B) U937 cells were seeded at equal ratio with DPSCs. The total number of U937 cells were counted at different times. (C) K562 cells were seeded at equal ratio with DPSCs. The total number of K562 cells were counted at different times.
Results
Increased Proliferation of Leukemia Cells with Direct Contact with DPSCs
We co-cultured HL-60 with DPSCs at 1:1 ratio (each, cells) in six-well tissue culture plates (Figure S2). At 24, 48 and 72 h, the cells were counted and compared with parallel cultures with HL-60 cells alone. The results showed significant () timeline increase in the proliferation of HL-60 cells with DPSCs, relative to HL-60 cells alone (Figure 1A). Similar studies with U937 cells showed an increase in proliferation, although delayed at 48 h with increase up to 72 h (Figure 1B). In contrast to HL-60 and U937 cells, K562 erythroleukemia cells showed no difference in proliferation, regardless of contacting DPSC (Figure 1C). In summary, this section showed a preference for DPSC with respect to enhanced proliferation of leukemia cell type. The data suggested responsiveness by HL-60 AML to DPSCs as compared to the myelomonocytic U937 cells.
Leukemia Cells Adhere to DPSCs
This let of studies analyzed the cultures to assess if the leukemia cells adhered to the DPSCs. We HL-60, K562 and U937 cells to 80% confluent DPSCs. At 2 and 12 h, we removed the non-adherent cells and then subjected the cultures to gentle washing to remove residual non-adherent cells. After this, the total number of adhered cells were counted. The results showed significant () increases in adherence by leukemia cells to DPSCs at 2 and 24 h (Figure 2B). HL-60 cell adherence was of particular interest since these cells proliferated in the presence of DPSCs (Figure 1). The total number of adhered HL-60 cells at the 12 h time point was calculated. The results indicated 5% HL-60 adhered to DPSCs (Figure 2C). In total, the three leukemia cell lines tested in this set of studies adhered to DPSCs.
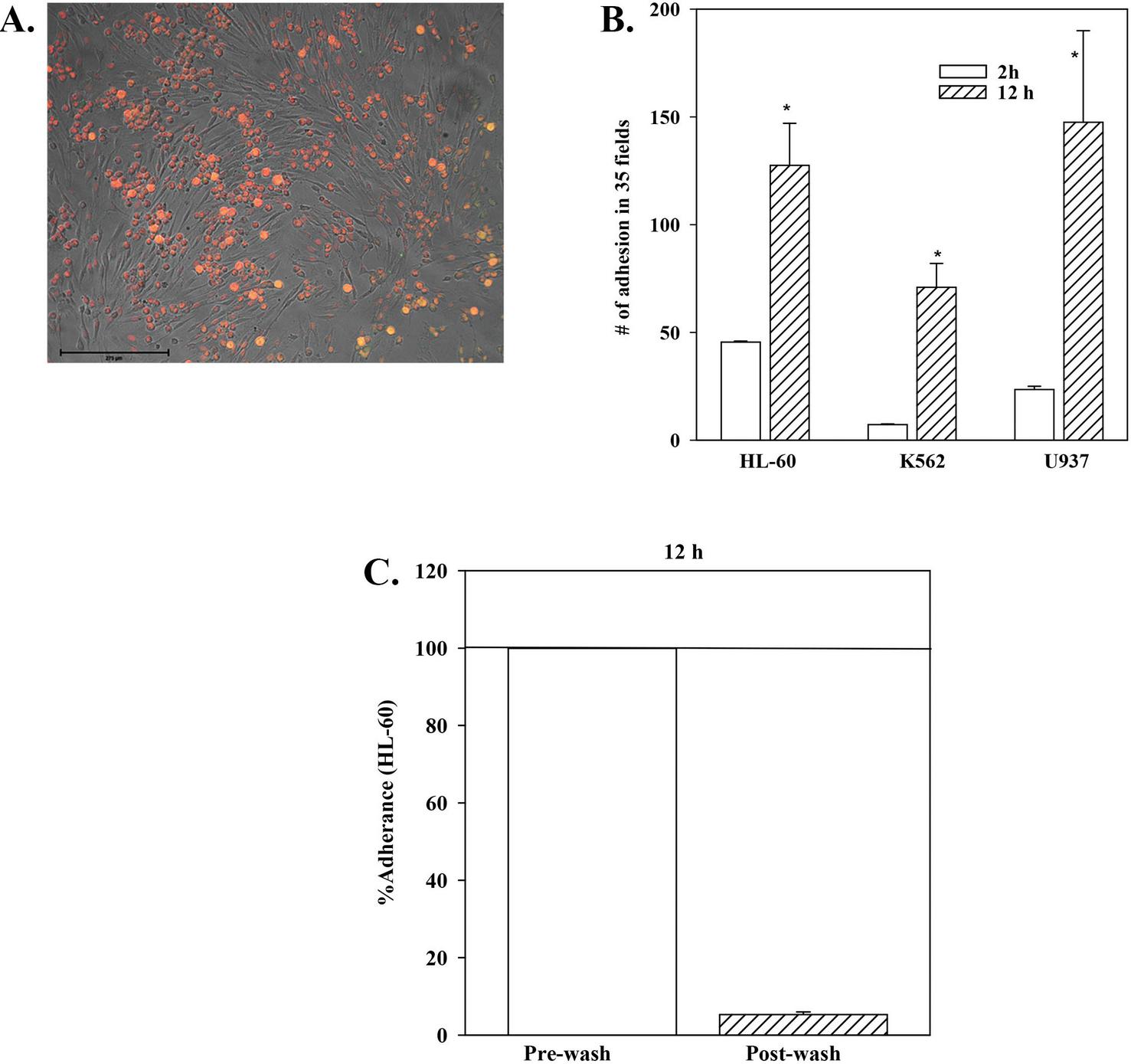
Figure 2 Adhesion between leukemia cells and DPSCSs. HL-60, K562 or U937 cells were added to DPSCs at 80% confluence. (A) An image used to count the total number of adhered leukemia cells. (B) The total number of adhered cells at 2 and 12 h are presented as the mean SD of three biological replicates. * p 0.05 vs. 2 h time point. (C) Percentage of adhered HL-60 cells. The data from “A” was used to calculate the percentage of HL-60 cells that adhered to DPSCs. The total number of cells at prewash were normalized as 100% and were used to calculate the adhered cells. The results are the mean SD of three biological replicates.
Intercellular Communication Between DPSCs and HL-60
Intercellular communication can occur by several methods such as tunnelling nanotubules (TNTs). We therefore assessed 48 h co-culture images for evidence of TNTs. As expected, adherence could occur when the HL-60 cells cluster on top of the DPSCs. Shown in Figure 3Ai is the inset of a representative image. These findings suggested a “tight” communication between the two cells. Of note is evidence of TNTs, which would suggest transfer of molecules from the DPSCs to the HL-60 cells (Figure 3Aii).
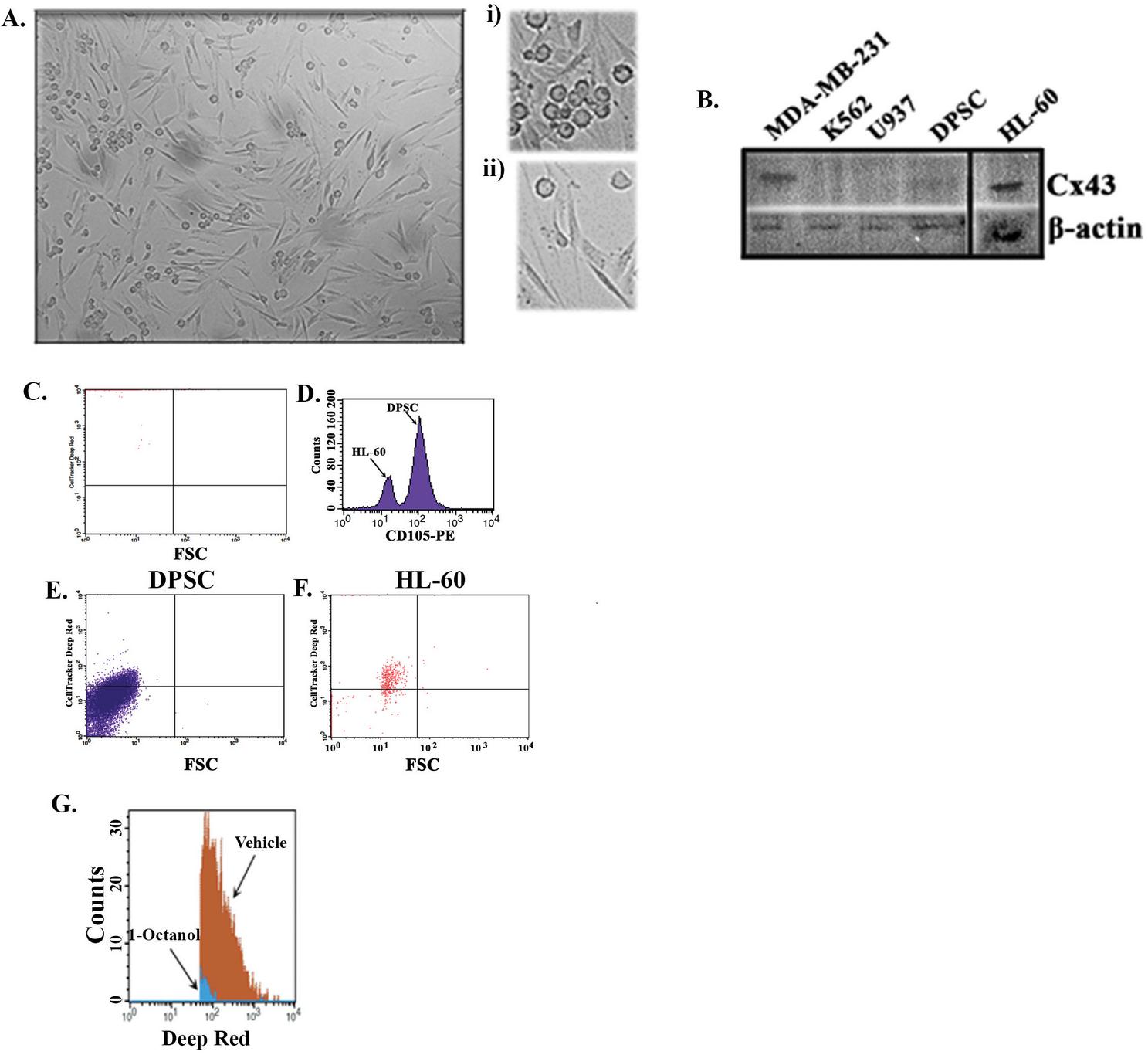
Figure 3 Intercellular communication between HL-60 cells and DPSCs. (A) Image of a representative field after 48 h of HL-60–DPSCs co-culture. The cells were seeded at 1:1 ratio. (i) Clusters of HL-60 on top of DPSC. (ii) Communication via TNT. (B) Western blot for Cx43 with extracts from MDA-MB-231 (positive control) [38], three leukemia cell lines and DPSC. The membrane was stripped and reprobed for -actin. The blot represents one of two biological analyses. (C)–(F) Dye transfer from DPSCs to HL-60 was conducted by labeling the DPSCs with deep red dye and then incubated with HL-60 cells at 1:1 ratio. Shown is undetectable deep red-labeled HL-60 at pre-incubation with the labeled DPSCs (C). The histogram showing the demarcation of HL-60 cells and DPSCs in co-culture based on positive staining for CD105 (D). Shown is deep red labeled DPSCs (E) and HL-60 cells after 24 h co-culture with HL-60 cells (F). (G) Representative histogram of reduced deep red in DPSC-HL-60 cells in co-culture with 1-Octanol or a vehicle.
Since the images in Figure 3A suggested intercellular communication between HL-60 and DPSCs, we asked if the HL-60 cells communicated with DPSCs via gap junctional intercellular communication (GJIC). First, we assessed the leukemia cells for Cx43 by western blot. The analysis included extracts from MDA-MB-231 breast cancer cells as positive control [38]. DPSCs were also analyzed for Cx43 since GJIC would require Cx43 expression in both the leukemia cells and DPSCs. The results showed a bright band for HL-60 and a lighter band with DPSC extract (Figure 3B). K562 and U937 showed light to undetectable bands (Figure 3B).
Since HL-60 cells showed a bright band for Cx43 in the western blot (Figure 3B), we asked if this could form GJIC. If so, there would be dye transfer from one cell to another. To this end, we labeled DPSCs with deep red and then incubated with unlabeled HL-60 cells at 1:1 ratio. The pre-incubated HL-60 cells showed no evidence of deep red (Figure 3C). After 24 h, we labeled the cells in co-cultures with anti-CD105-PE and determined that this labeling could demarcate DPSCs and HL-60 cells (Figure 3D). After gating the DPSCs, we showed reduced deep red in this population but bright fluorescence in the HL-60 cells (Figures 3E and 3F). This indicated transfer of deep red from DPSCs to HL-60 cells. Next, the co-cultures were repeated with 1-Octanol or a vehicle. The addition of 1-octanol significantly decreased the transfer of deep red into HL-60 cells. This section demonstrated intracellular communication between DPSCs and HL-60 cells through GJIC.
Dye Transfer in Non-adherent HL-60 Cells
In this section, we ask if the dye was transferred to HL-60 cells by methods other than GJIC. This question was relevant since we noted evidence of potential communication by TNT (Figure 3A). We therefore quantitated the percentages of deep red positive HL-60 in suspension and those that adhered to the DPSCs. Flow cytometry for deep red indicated dye transfer in the adherent and non-adherent HL-60 cells (Figure 4A). The data indicated about 19% positive HL-60 cells within the adherent population and 33% positive in suspension (Figure 4B).
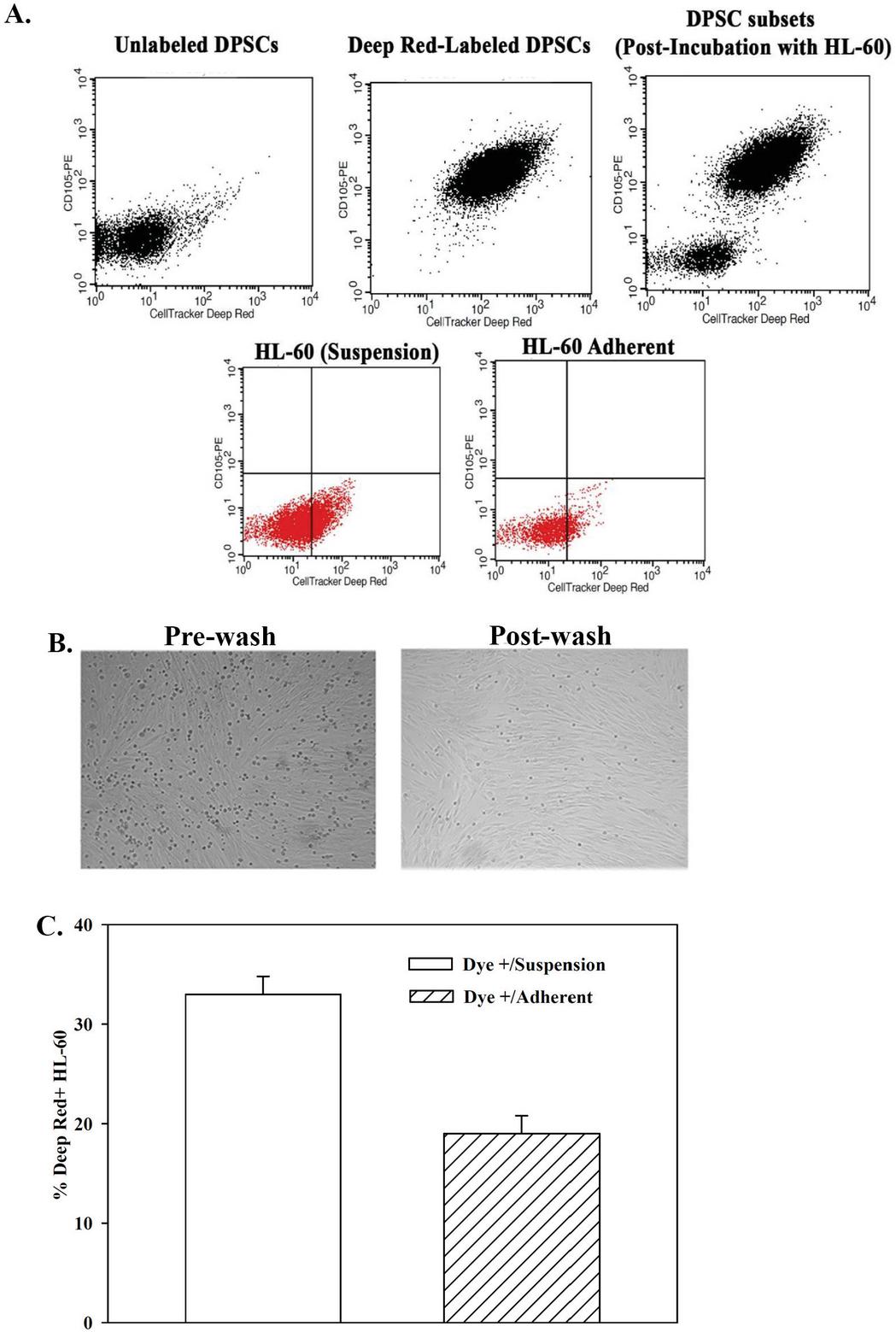
Figure 4 Deep red in adherent and non-adherent HL-60 in co-culture with DEPC. (A) HL-60 and DPSCs were co-cultured at 1:1 ratio. The DPSCs were labeled with deep red dye. After 16 h, the suspension cells were collected. The adherent cells were washed and then collected after trypsinization. The latter cells were labeled for CD105 to demarcate DPSCs. Top left panel: Unlabeled DPSCs alone. Top middle panel: Deep red labeled DPSCs alone. Top right panel: DPSCs after co-culture. Lower left panel: HL-60 cells in media (suspension). Lower right: HL-60 cells that adhere to DPSCs after co-culture. (B) Panels show representative co-cultures before and after washes. The images were used to quantitate the number of dye-labeled HL-60 cells in the adherent and non-adherent population were quantitated from three biological replicates. (C) The data from “B” are shown in the graph with the mean SD percentage of total deep red positive HL-60 cells in suspension and adherence to DPSCs on the Y-axis.
Preference of an HL-60 Subset to Establish GJIC with DPSC
Previous studies indicated that the SORE6, which is under the expression of tandem repeats of Oct4 and Sox 2 could demarcate glioblastoma stem cells [36]. In this set of studies, we asked if this lentivirus can determine if a specific subset of HL-60 cells showed preference for intercellular communication, using dye transfer as an indicator.
The stable SORE6 HL-60 cells were stratified based on fluorescence intensity. The cells were added to co-cultures and then labeled with CD105 to eliminate analysis of the DPSCs, which were labeled with deep red (Figure 5A). The HL60 subsets were stratified as high (hi), medium (med) and low (lo), based on fluorescence intensities after 24 h in co-culture (Figures 5B). After 24 h, the adherent and non-adherent HL-60 subsets were assessed for dye transfer (Figure 5C). The hi and med HL-60 subsets show 2% dye transfer within the adherent and suspension cells whereas the lo subset shows high dye transfer. In summary, the different subsets of SORE6 HL-60 cells could interact with DPSCs as evidenced by dye transfer. Interestingly, the cells with the lowest SORE 6 expression were more responsive to the dye transfer.
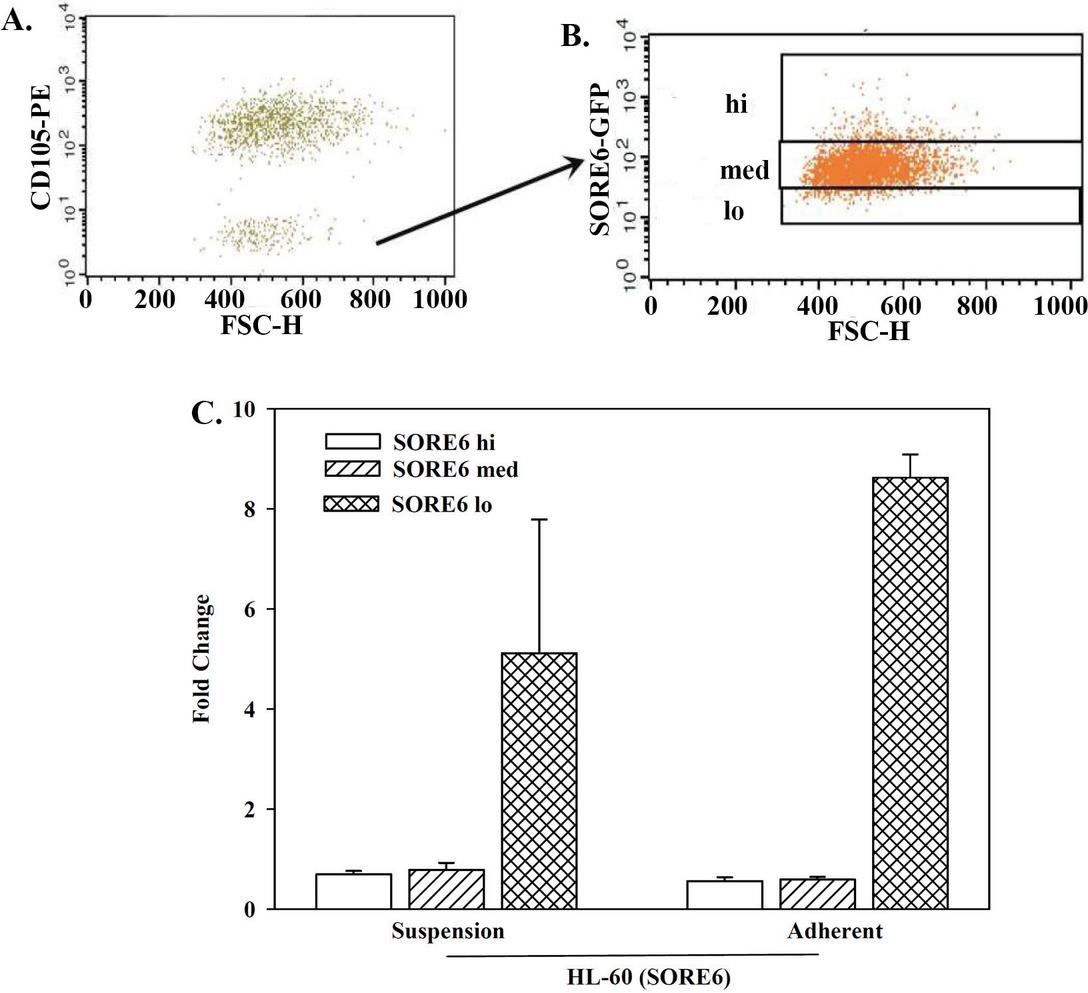
Figure 5 Dye transfer between DPSCs and HL-60 subsets. SORE6-HL-60 cells were sorted as hi, med and lo. The sorted cells were added to 80% confluent DPSCs and the cells in suspension and those that adhere to DPSCs were evaluated for dye transfer by flow cytometry. (A) Figure shows how HL-60 (CD105-) and DPSCs (CD105+) were demarcated. (B) Shows how the HL-60 cells from “B” were stratified for SORE6 hi, med and low. (C) Three biological replicates from (A) and (B) are presented as the mean SD of dye within the adherent and non-adherent/suspension HL-60 subsets.
Discussion
The identification of leukemia in the oral cavity warrants research to dissect the mechanisms by which the oral cavity supports hematological tumors. The literature has reported several methods by which AML risk factors have been suggested [39, 40]. However, there is no identifiable risk factor for most AML cases. Therefore, it is important to identify new diagnostic, prognostic and therapeutic approaches, such as the oral cavity as an extramedullary site of AML. The question is, does AML exist as dormant cancer cells in the oral tissue? Can AML develop simultaneously in the oral tissue and bone marrow? The expectation is that treating AML would improve the intraoral clinical presentation. However, is it possible that residual AML cells remain in the oral tissue and give rise to relapse/metastasis? Investigations into the mechanisms by which tumor cells survive and the method of integration in the oral cavity remains a poor area of research. This article focused on research on three hematological tumor cell lines – AML HL-60, erythroleukemia K562 and myelomonocytic U937.
HL-60 has shown enhanced proliferation in the presence of DPSCs. Although U937 showed similar proliferation, the proliferative response was reduced when compared to the response by HL-60 cells. As expected, K562 cultured alone showed timeline proliferation. However, when K562 cells were placed in contact with DPSCs, there was no change in proliferation. However, there was a delayed trend in which DPSCs increased the proliferation of K562 (Figure 1). These varied responses suggest that the role of DPSCs might be leukemia specific and/or subset mediated. However, it is difficult to make a definite statement since only one cell line was used from each leukemia type. Indeed, this first set of studies did not apply an experimental model to recapitulate the oral cavity since only DPSCs were added to the cultures. A true recapitulation would include other microenvironmental cell types in the oral cavity, including blood vessels and nerve fibers.
Ideally, it would be efficient to include a model that represents the oral cavity in 3D. This could be achieved with tissue organoids, in vivo studies, and/or 3D bioprinting. In the case of the leukemia cells, such studies should compare the with hematological models in which the leukemia cells are evaluated in the BM. Our laboratory has developed an efficient bioprinting model to recapitulate the BM that could be used in parallel with 3D models of the oral cavity [41, 42].
It is generally assumed that cells would de-adhere during proliferation. However, the data indicated that increased proliferation did not affect adherence of the three cell lines to DPSCs. This was interesting when considering that cancer cell subsets seem to prefer specific type of communication with healthy mesenchymal stem cells [43]. In this regard, the proliferating leukemia cells that adhere could be in cellular quiescence. The premise for proposing cellular quiescence of the HL-60 cells is based on other studies that reported about 5% of cancer stem cells in breast cancer [44].
Combining leukemia cell proliferation and adherence to DPSCs, led to several possible methods of communication. The data showed evidence of TNTs as a method of communication. However, this study observed the co-cultures up to 72 h. Therefore, current evidence of TNT would be at the beginning of intercellular communication. It is possible that longer co-culture could define if TNTs might be a stronger method of intercellular communication. Future studies are needed to address TNTs that are linked to intercellular communication in the cancer mechanism [45, 46]. The need to follow up on TNTs is due to reports showing the tubules transfering mitochondria to other cells, which could alter the behavior of cancer cells [47].
In another method of communication, the experimental evidence indicated GJIC between HL-60 cells and DPSCs. The evidence of GJIC was verified by the pharmacological agent 1-octanol. Other studies are required to further investigate the findings on GJIC. Our studies on GJIC focused on HL-60 due to the noted 5% adherence to DPSCs. Our laboratory previously reported on GJIC between breast cancer cells and bone marrow-derived mesenchymal stem cells and macrophages [38, 43, 46, 48, 49]. We noted Cx43 protein with whole cell extracts from HL-60, K562 and U937. Since Cx43 was also noted in DPSCs, there is likely intercellular communication by the formation of cellular junction. Analyses of the individual cells indicated bright bands for Cx43 with HL-60 extract and a light band with DPSCs extract (Figure 3). It is possible that other connexins were involved in GJIC. Additionally, upon contact, the Cx43 expression might increase. This question will require further studies since answers to this question will be insightful to understand how the cells communicate to support the survival of leukemia in the oral cavity. Additional studies will require more targeted elimination of connexin with knockdown or knockout studies [43].
Since SORE-6 as well as the full Oct4a regulatory region were able to demarcate a cancer cell subset, we employed SORE-6 transfected HL-60 to study how the different subset interact with DPSCs [36, 38]. In this study, a similar method was applied to determine what subsets might be responsible to form GJIC with SORE6 HL-60 cells. The fluorescence intensity of SORE6 transfectants is proportional to the stemness of the cell [36, 50, 51]. We compared SORE6 HL-60 hi, med and low subsets for dye transfer from deep red labeled DPSCs. At 24 h, the hi and med HL-60 subsets show 2% dye transfer within the adherent and suspension cells whereas the lo subset shows high dye transfer. This was a surprising finding since other studies showed a direct relationship between GJIC and the subset with the most intense SORE6. Based on this, we deduced that there must be other methods of communication between HL-60 cells and DPSCs. These are ongoing studies by our group.
At this time, similarities and/or differences in the mechanisms by which AML interacts with DPSCs and bone marrow mesenchymal stem cells need to be addressed to determine if the interaction differs and, if so, how this would affect the current treatment. To understand how AML or any other hematological malignancy can survive in the oral cavity, one must think of the literature pertaining to hematopoiesis in the oral cavity. The reports indicated a small and steady population of circulating hematopoietic stem and progenitor cells (HSPCs) at extramedullary sites, such as gingiva [52]. The growing evidence indicated hematopoietic activity in healthy gingiva [53]. Thus, there is a possibility that, as one ages, the otherwise healthy gingival HSPCs could transform to AML. The other possibility is that AML from the bone marrow could migrate to the gingiva. The fundamental question is to initiate research to understand how AML cells survive in the oral tissue. In this regard, this study provides insights that open pathways to determine the role of the oral cavity in AML pathology.
In summary, this article discusses a gap in knowledge with respect to the mechanisms by which AML survives and develops in the oral cavity. Specifically, interactions between these tumor cells and intraoral niche for the cancer cells to survive and perhaps to resist current drug treatments. To the best of our knowledge, there have been no studies to address the oral tissue as a potential site for cancer dormancy and to protect current treatment. To this end, this report forms the basis for future studies to begin a path to determine if current AML and other leukemia treatments are effective in the oral cavity.
References
[1] Janowiak-Majeranowska, A., J. Osowski, B. Mikaszewski, and A. Majeranowski. 2022. Secondary oral cancer after systemic treatment of hematological malignancies and oral GVHD: a systematic review. Cancers 14: 2175.
[2] Hirshberg, A., A. Shnaiderman-Shapiro, I. Kaplan, and R. Berger. 2008. Metastatic tumours to the oral cavity - pathogenesis and analysis of 673 cases. Oral Oncol 44: 743–752.
[3] Anderson, L. J., M. Girguis, E. Kim, J. Shewale, M. Braunlin, W. Werther, J. E. Hidalgo-Lopez, F. Zaman, and C. Kim. 2024. A temporal and multinational assessment of acute myeloid leukemia (AML) cancer incidence, survival, and disease burden. Leuk Lymphoma: 1–10.
[4] Gopal, S., K. P. Shetty, V. Jindal, and M. Saritha. 2011. Interrelationship of Endodontic-Periodontal Lesions-An Overview. Indian J Dental Sci 3.
[5] Bruno, K. F., F. B. Barletta, W. T. Felippe, J. A. Silva, A. H. G. de Alencar, and C. Estrela. 2014. Oxygen saturation in the dental pulp of permanent teeth: a critical review. Journal of endodontics 40: 1054–1057.
[6] Fernandes, K. S., M. Gallottini, T. Castro, M. F. Amato, J. S. Lago, and P. H. Braz-Silva. 2018. Gingival leukemic infiltration as the first manifestation of acute myeloid leukemia. Special Care in Dentistry 38: 160–162.
[7] Sonoi, N., Y. Soga, H. Maeda, K. Ichimura, T. Yoshino, K. Aoyama, N. Fujii, Y. Maeda, M. Tanimoto, R. Logan, J. Raber-Durlacher, and S. Takashiba. 2012. Histological and immunohistochemical features of gingival enlargement in a patient with AML. Odontology 100: 254–257.
[8] Shankarapillai, R., M. A. Nair, R. George, and L. J. Walsh. 2010. Periodontal and gingival parameters in young adults with acute myeloid leukaemia in Kerala, South India. Oral Health Prev Dent 8: 395–400.
[9] Gallipoli, P., and M. Leach. 2007. Gingival infiltration in acute monoblastic leukaemia. Br Dent J 203: 507–509.
[10] Demirer, S., H. Ozdemir, M. Sencan, and I. Marakoglu. 2007. Gingival hyperplasia as an early diagnostic oral manifestation in acute monocytic leukemia: a case report. Eur J Dent 1: 111–114.
[11] Cooper, C. L., R. Loewen, and T. Shore. 2000. Gingival hyperplasia complicating acute myelomonocytic leukemia. J Can Dent Assoc 66: 78–79.
[12] Hasan, S., N. I. Khan, and L. B. Reddy. 2015. Leukemic gingival enlargement: Report of a rare case with review of literature. Int J Appl Basic Med Res 5: 65–67.
[13] Hou, G. L., and C. C. Tsai. 1988. Primary gingival enlargement as a diagnostic indicator in acute myelomonocytic leukemia. A case report. J Periodontol 59: 852–855.
[14] Arnold, M., E. Morgan, H. Rumgay, A. Mafra, D. Singh, M. Laversanne, J. Vignat, J. R. Gralow, F. Cardoso, and S. Siesling. 2022. Current and future burden of breast cancer: Global statistics for 2020 and 2040. The Breast 66: 15–23.
[15] Cammarata-Scalisi, F., K. Girardi, L. Strocchio, P. Merli, A. Garret-Bernardin, A. Galeotti, F. Magliarditi, A. Inserra, and M. Callea. 2020. Oral Manifestations and Complications in Childhood Acute Myeloid Leukemia. Cancers (Basel) 12.
[16] Dreizen, S., K. B. McCredie, M. J. Keating, and M. A. Luna. 1983. Malignant gingival and skin “infiltrates” in adult leukemia. Oral Surg Oral Med Oral Pathol 55: 572–579.
[17] Pugh, C. W., and P. J. Ratcliffe. 2003. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9: 677–684.
[18] Forte, D., M. García-Fernández, A. Sánchez-Aguilera, V. Stavropoulou, C. Fielding, D. Martín-Pérez, J. A. López, A. S. H. Costa, L. Tronci, E. Nikitopoulou, M. Barber, P. Gallipoli, L. Marando, C. L. Fernández de Castillejo, A. Tzankov, S. Dietmann, M. Cavo, L. Catani, A. Curti, J. Vázquez, C. Frezza, B. J. Huntly, J. Schwaller, and S. Méndez-Ferrer. 2020. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape from Chemotherapy. Cell Metab 32: 829–843.e829.
[19] Williams WJ, B. E., Erslev AJ, Lichtman MA. 1990. Hematology. 4th ed. M. Hill, ed, New York. 243–244.
[20] Felix, D. E., and J. Lukens. 1986. Oral symptoms as a chief sign of acute monoblastic leukemia: report of case. J Am Dent Assoc 113: 899–900.
[21] Brenneise, C. V., J. S. Mattson, and J. R. Commers. 1988. Acute myelomonocytic leukemia with oral manifestations: report of case. J Am Dent Assoc 117: 835–837.
[22] Epstein, J. B., R. W. Priddy, T. Sparling, and L. Wadsworth. 1986. Oral manifestations in myelodysplastic syndrome. Review of the literature and report of a case. Oral Surg Oral Med Oral Pathol 61: 466–470.
[23] Khera, P., M. J. Zirwas, and J. C. English, 3rd. 2005. Diffuse gingival enlargement. J Am Acad Dermatol 52: 491–499.
[24] Pogrel, M. A. 1978. Acute leukemia. An atypical case presenting with gingival manifestations. Int J Oral Surg 7: 119–122.
[25] McKenna, S. J. 2000. Leukemia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 89: 137–139.
[26] Wu, J., J. E. Fantasia, and R. Kaplan. 2002. Oral manifestations of acute myelomonocytic leukemia: a case report and review of the classification of leukemias. J Periodontol 73: 664–668.
[27] Elad, S., J. E. Raber-Durlacher, M. T. Brennan, D. P. Saunders, A. P. Mank, Y. Zadik, B. Quinn, J. B. Epstein, N. M. Blijlevens, and T. Waltimo. 2015. Basic oral care for hematology–oncology patients and hematopoietic stem cell transplantation recipients: a position paper from the joint task force of the Multinational Association of Supportive Care in Cancer/International Society of Oral Oncology (MASCC/ISOO) and the European Society for Blood and Marrow Transplantation (EBMT). Supportive Care in Cancer 23: 223–236.
[28] Menezes, L., and J. R. Rao. 2012. Acute myelomonocytic leukemia presenting with gingival enlargement as the only clinical manifestation. J Indian Soc Periodontol 16: 597–601.
[29] Sollecito, T. P., J. Draznin, E. Parisi, K. Duffy, E. A. Stadtmauer, S. M. Luger, S. J. Schuster, D. Tsai, and D. L. Porter. 2003. Leukemic gingival infiltrate as an indicator of chemotherapeutic failure following monoclonal antibody therapy: a case report. Spec Care Dentist 23: 108–110.
[30] Chen, L., H. M. Linden, B. O. Anderson, and C. I. Li. 2014. Trends in 5-year survival rates among breast cancer patients by hormone receptor status and stage. Breast Cancer Res Treatment 147: 609–616.
[31] Rizvanov, A. A., M. E. Yalvac, A. K. Shafigullina, Salafutdinov, II, N. L. Blatt, F. Sahin, A. P. Kiyasov, and A. Palotas. 2010. Interaction and self-organization of human mesenchymal stem cells and neuro-blastoma SH-SY5Y cells under co-culture conditions: A novel system for modeling cancer cell micro-environment. Eur J Pharm Biopharm 76: 253–259.
[32] Dogan, A., S. Demirci, H. Apdik, E. A. Apdik, and F. Sahin. 2017. Dental pulp stem cells (DPSCs) increase prostate cancer cell proliferation and migration under in vitro conditions. Tissue Cell 49: 711–718.
[33] Raj, A. T., S. Kheur, R. Bhonde, V. R. Mani, H. A. Baeshen, and S. Patil. 2021. Assessing the effect of human dental pulp mesenchymal stem cell secretome on human oral, breast, and melanoma cancer cell lines. Saudi J Biol Sci 28: 6556–6567.
[34] Shi, S., and S. Gronthos. 2003. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Mineral Res 18: 696–704.
[35] Izadirad, M., Z. Huang, F. Jafari, A. A. Hamidieh, A. Gharehbaghian, Y.-D. Li, L. Jafari, and Z.-S. Chen. 2021. Extracellular vesicles in acute leukemia: A mesmerizing journey with a focus on transferred microRNAs. Frontiers Cell Dev Biol 9: 766371.
[36] Savanur, V. H., A. Sarkar, A. Petryna, K. Nguyen, J. Benites-Sandoval, M. Gergues, A. Hatefi, and P. Rameshwar. 2024. A Platform to Establish a Working Hierarchy of Glioblastoma Multiforme Cells:: Implication on Cancer Cell-microenvironment Interaction and Response to Drugs. Intl J Transl Sci: 177–200.
[37] Tsolaki, I., D. Rojas, A. Eljarrah, and P. Rameshwar. 2024. Isolating Dental Pulp Stem Cells. In Stepwise Culture of Human Adult Stem Cells. River Publishers. 45–52.
[38] Patel, J. P., M. Gönen, M. E. Figueroa, H. Fernandez, Z. Sun, J. Racevskis, P. Van Vlierberghe, I. Dolgalev, S. Thomas, O. Aminova, K. Huberman, J. Cheng, A. Viale, N. D. Socci, A. Heguy, A. Cherry, G. Vance, R. R. Higgins, R. P. Ketterling, R. E. Gallagher, M. Litzow, M. R. van den Brink, H. M. Lazarus, J. M. Rowe, S. Luger, A. Ferrando, E. Paietta, M. S. Tallman, A. Melnick, O. Abdel-Wahab, and R. L. Levine. 2012. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366: 1079–1089.
[39] Tallman, M. S., E. S. Wang, J. K. Altman, F. R. Appelbaum, V. R. Bhatt, D. Bixby, S. E. Coutre, M. De Lima, A. T. Fathi, M. Fiorella, J. M. Foran, A. C. Hall, M. Jacoby, J. Lancet, T. W. LeBlanc, G. Mannis, G. Marcucci, M. G. Martin, A. Mims, M. R. O’Donnell, R. Olin, D. Peker, A. Perl, D. A. Pollyea, K. Pratz, T. Prebet, F. Ravandi, P. J. Shami, R. M. Stone, S. A. Strickland, M. Wieduwilt, K. M. Gregory, L. Hammond, and N. Ogba. 2019. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Net 17: 721–749.
[40] Arber, D. A., A. Orazi, R. Hasserjian, J. Thiele, M. J. Borowitz, M. M. Le Beau, C. D. Bloomfield, M. Cazzola, and J. W. Vardiman. 2016. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127: 2391–2405.
[41] Moore, C. A., Z. Siddiqui, G. J. Carney, Y. Naaldijk, K. Guiro, A. I. Ferrer, L. S. Sherman, M. Guvendiren, V. A. Kumar, and P. Rameshwar. 2021. A 3D Bioprinted Material That Recapitulates the Perivascular Bone Marrow Structure for Sustained Hematopoietic and Cancer Models. Polymers (Basel) 13.
[42] Castillo, M., K. Liu, L. Bonilla, and P. Rameshwar. 2007. The immune properties of mesenchymal stem cells. Int J Biomed Sci 3: 76–80.
[43] Sinha, G., A. I. Ferrer, S. Ayer, M. H. El-Far, S. H. Pamarthi, Y. Naaldijk, P. Barak, O. A. Sandiford, B. M. Bibber, G. Yehia, S. J. Greco, J. G. Jiang, M. Bryan, R. Kumar, N. M. Ponzio, J. P. Etchegaray, and P. Rameshwar. 2021. Specific N-cadherin-dependent pathways drive human breast cancer dormancy in bone marrow. Life Sci Alliance 4:e202000969.
[44] Patel, S. A., S. H. Ramkissoon, M. Bryan, L. F. Pliner, G. Dontu, P. S. Patel, S. Amiri, S. R. Pine, and P. Rameshwar. 2012. Delineation of breast cancer cell hierarchy identifies the subset responsible for dormancy. Sci Rep 2: 906.
[45] Dubois, F., M. Bénard, B. Jean-Jacques, D. Schapman, H. Roberge, A. Lebon, D. Goux, B. Monterroso, N. Elie, and H. Komuro. 2020. Investigating tunneling nanotubes in cancer cells: Guidelines for structural and functional studies through cell imaging. BioMed Res Intl 2020: 2701345.
[46] Patel, S. A., M. A. Dave, S. A. Bliss, A. B. Giec-Ujda, M. Bryan, L. F. Pliner, and P. Rameshwar. 2014. Treg/Th17 polarization by distinct subsets of breast cancer cells is dictated by the interaction with mesenchymal stem cells. J Canccr Stem Cell Res 2014.
[47] Guerra, F., A. A. Arbini, and L. Moro. 2017. Mitochondria and cancer chemoresistance. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1858: 686–699.
[48] Greco, S. J., S. Ayer, K. Guiro, G. Sinha, R. J. Donnelly, M. H. El-Far, L. S. Sherman, Y. Kenfack, S. H. Pamarthi, M. Gergues, O. A. Sandiford, M. J. Schonning, J. P. Etchegaray, and P. Rameshwar. 2021. Restoration of aged hematopoietic cells by their young counterparts through instructive microvesicles release. Aging (Albany NY) 13: 23981–24016.
[49] DiNardo, C. D., and A. H. Wei. 2020. How I treat acute myeloid leukemia in the era of new drugs. Blood 135: 85–96.
[50] Tang, B., A. Raviv, D. Esposito, K. C. Flanders, C. Daniel, B. T. Nghiem, S. Garfield, L. Lim, P. Mannan, A. I. Robles, W. I. Smith, Jr., J. Zimmerberg, R. Ravin, and L. M. Wakefield. 2015. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Reports 4: 155–169.
[51] Tang, B., A. Raviv, D. Esposito, K. C. Flanders, C. Daniel, B. T. Nghiem, S. Garfield, L. Lim, P. Mannan, and A. I. Robles. 2015. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Reports 4: 155–169.
[52] Massberg, S., P. Schaerli, I. Knezevic-Maramica, M. Köllnberger, N. Tubo, E. A. Moseman, I. V. Huff, T. Junt, A. J. Wagers, I. B. Mazo, and U. H. von Andrian. 2007. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 131: 994–1008.
[53] Bergmann, O. J., H. P. Philipsen, and J. Ellegaard. 1988. Isolated gingival relapse in acute myeloid leukaemia. Eur J Haematol 40: 473–476.
International Journal of Translational Science, Vol. 1, 91–112.
doi: 10.13052/ijts2246-8765.2025.005
© 2025 River Publishers