Extracellular Vesicles from Healthy Placenta Stem Cells Restored the Immune Licensing Function of Preeclamptic Placenta Stem Cells
Jessica T. Greenberg1, Lauren S. Sherman2, Shauna F. Williams1,* and Pranela Rameshwar2,*
1Department of Obstetrics, Gynecology, and Reproductive Health, Rutgers New Jersey Medical School, Newark, NJ, United States
2Department of Medicine Rutgers New Jersey Medical School, Newark, NJ, United States
E-mail: williash@njms.rutgers.edu; rameshwa@njms.rutgers.edu
*Corresponding Author
Received 14 August 2025; Accepted 03 September 2025
Abstract
Preeclampsia (PE) contributes to pregnancy-related morbidity and mortality, with enhanced inflammation. Healthy placenta stem cells (P-MSCs) can be licensed into immune suppressor cells to mitigate inflammation. Since PE is associated with inflammation, the question is why the associated P-MSCs cannot suppress the response. PE P-MSCs have been shown to be dysfunctional with respect to decreased anti-inflammatory response, cell cycle dysregulation, and reduced production of immune suppressive cytokines. Aspirin (ASA) treatment partly reversed these dysfunctions via epigenetic reprogramming. We tested the hypothesis that extracellular vesicles (EVs) from healthy P-MSC could reset PE P-MSCs to a healthy phenotype, including cell cycle dysregulation and anti-inflammatory licensing. EVs from healthy MSCs were collected and the number of particles quantified. The isolated EVs were added to PE P-MSCs. Control cultures treated the PE P-MSCs with 1 mM ASA. The treated cells were assessed for the epigene regulator TDG and cell cycle linked CDK4, p21, and p53 by western blot, or assessed as third-party suppression in a one-way mixed lymphocyte reaction (MLR). EV- and ASA-treated PE P-MSC suppressed MLR, similar to healthy P-MSCs. However, an evaluation of p21, CDK4, p53, and TDG suggested that EVs impart a more stable restoration of PE P-MSCs when exposed to healthy EVs. This study provides insights into the method by which healthy P-MSCs can function to restore PE P-MSCs, and in vivo microenvironmental restoration.
Keywords: Stem cells, placenta, preeclampsia, aspirin, exosomes, extracellular vesicles, cell cycle.
Key Points
Exosomes from healthy placenta stem cells (P-MSCs) can restore the ability of preeclamptic P-MSC to be licensed as immune suppressor cells.
Exosomes from healthy P-MSCs altered the expression of cell cycle and DNA repair proteins, and this depended on the number of particles.
Exosomes from healthy P-MSCs might recapitulate the effects of aspirin.
Introduction
Preeclampsia (PE), a hypertensive disorder of pregnancy, is a major contributor to pregnancy-related morbidity and mortality [1–4]. PE can lead to maternal seizures, stroke, end organ damage, placental abruption, fetal growth restriction, oligohydramnios, non-reassuring fetal status, preterm birth, and stillbirth [5]. PE pathophysiology is not completely elucidated [5], but inflammation appears to be a critical factor [6, 7]. A leading theory suggests that suboptimal trophoblastic invasion and spiral artery remodeling leads to inflammation, endothelial damage, and platelet aggregation. These effects could lead to placental infarcts, resulting in placental insufficiency and downstream PE-associated effects [8]. Aspirin (ASA), with its anti-inflammatory and anti-platelet activity, has become the standard of care to prevent PE [9, 10]. However, despite the use of ASA, the prevalence of PE remains high [1].
Mesenchymal stem cells (MSCs) can be found in adult and fetal tissues, such as bone marrow, adipose tissues, and placenta (P-MSCs). MSCs can repair tissue damage and exert both immune suppressor and inflammatory functions [11]. These dual functions depended on the inflammatory milieu with immune suppression within increased inflammation [12, 13]. Our group has previously reported on anti-inflammatory dysfunction by P-MSCs from PE and epigenetic changes [14].
ASA treatment extended the quiescent phase of P-MSCs from PE, which would provide greater opportunity for DNA repair [14]. This partially occurred by increases in p21, p53, and TGF1, and other cell cycle associated proteins with inhibition of other cyclin associated genes such as Cdk4 [14]. These changes correlated with an increased base excision repair (BER) pathway, suggesting that ASA treatment could induce genomic stability of PE P-MSCs with [15] reduced DNA damage. Similarly, nucleotide excision repair (NER) expression decreased four-fold after ASA treatment. Functionally, ASA treatment restored the ability of PE P-MSC to be licensed with anti-inflammatory function, with concomitant increase in active TGF-1 [14, 16].
MSCs within a tissue microenvironment can establish intercellular communication by soluble and insoluble factors. Among these factors are extracellular vesicles (EVs) with protein, DNA, mRNA, and lipid cargo [11]. Exosomes are EVs and are 30–150 nm in size. We have reported on the ability of EVs from young hematopoietic cells to restore the function of aged hematopoietic cells [17]. This restorative function might occur endogenously to maintain tissue homeostasis. To this end, we propose that EVs from healthy P-MSCs would be able to restore the dysfunction of PE-derived P-MSCs.
EVs from umbilical cord MSCs can enhance the invasion and migration of trophoblasts, inhibit trophoblast apoptosis, and improve symptoms of PE in rat models [18–23]. EVs from MSCs can exert anti-inflammatory effects and this correlated with increased production of anti-inflammatory cytokines [24–27]. This study reported on immune reset when EVs from healthy P-MSCs were added to PE P-MSCs. Specifically, we reported on EV treated PE P-MSCs to suppress the immune response in a mixed lymphocyte reaction. We also showed stable reset in cell cycle and DNA proteins related to low dose aspirin. The clinical impact of these findings is discussed.
Materials and Methods
Reagents and Antibodies
Phosphate buffered saline (PBS), Dulbecco’s Modified Eagle Medium–high glucose (DMEM), fetal bovine serum (FBS), penicillin/streptomycin (P-S), L-glutamine, bovine serum albumin (BSA), aspirin (acetylsalicylic acid, ASA), Ficoll Hypaque, and protease inhibitor were purchased from Sigma-Aldrich (St. Louis, MO); Trypsin was purchased from Life Technologies (Carlsbad, CA); Precision Plus Protein Kaleidoscope Ladder, Bradford protein assay and Laemmli buffer from Bio-Rad (Hercules, CA); rabbit anti-Alix (1:000), rabbit anti-Acetyl Histone 3 (1:1000), rabbit anti-p53 (1:1000), and murine anti-p21 (1:1000) were purchased from Cell Signaling Technology (Danvers, MA); murine anti-CDK4 (1:250 from Thermo Fischer Scientific (Waltham, MA; murine anti-TDG (1:5000) from Abcam (Cambridge, MA); HRP conjugated anti-mouse and anti-rabbit IgG, mammalian protein extraction reagent (MPER), stripping buffer, chemiluminescent test (ECL), and adipocyte induction media from Thermo Fischer Scientific (Waltham, MA); osteocyte induction media purchased from StemPro-Life Technologies (Grand Island, NY); APC conjugated murine anti-CD29, -CD90, PE conjugated murine anti-CD45 and PerCP-Cv5.5 conjugated anti-CD105 from BD Technologies (Durham, NC).
Human Subjects
This study was approved by Rutger Institutional Review Board. All donors signed the informed consent. PE was a clinical diagnosis determined by the treating team, as defined by ACOG. Healthy pregnancies had no placental dysfunction, intraamniotic infection, hypertension, diabetes, HIV or Hepatitis B or Hepatitis C. Placentas were collected from healthy and PE singleton pregnancies delivered at- or near-term. All pregnancies were negative for other reported complications. Peripheral blood from healthy donors (20–35 yrs) were also collected. P-MSCs used were from an ongoing collected repository.
Table 1 outlines the demographics of three additional subjects that were not previously described. In addition to use of P-MSCs from the samples shown in Table 1, P-MSCs were also used from cryopreserved cells. Donors of the stored P-MSCs were were previously described [14–16].
Table 1 Demographics/obstetrical and neonatal information
| Specific Data | Patient 1 | Patient 2 | Patient 3 |
| Diagnosis of PE with severe features | Yes | No | No |
| Age (years) | 23 | 30 | 23 |
| Gravidity | 2 | 5 | 2 |
| Parity | 0 | 3 | 1 |
| Gestational age at delivery (days) | 275 | 277 | 277 |
| BMI at delivery | 36.3 | 28.4 | 28.2 |
| Neonatal gender | Male | Male | Male |
| Neonatal birth weight (g) | 3510 | 3145 | 3100 |
| Apgar at 1 minute | 9 | 9 | 8 |
| Apgar at 5 minutes | 9 | 9 | 9 |
| Maximum systolic BP (mmHg) | 138 | 134 | 129 |
| Maximum diastolic BP (mmHg) | 110 | 84 | 87 |
These three donors are in addition to the use of other P-MSCs that were previously reported [14–16].
P-MSC Isolation and Culture
Placentas were collected and immediately dissected under the laminar flow hood, as described in [14]. The chorionic membrane was separated from the amniotic membrane using sharp dissection followed by cutting into 3–5 mm pieces. The recovered tissues were added into tissue culture treated plates. The plates were incubated for 10 mins at room temperature. After this, DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% L-glutamine (MSC media) were added to the plates followed by immediate transfer to an incubator – 37∘C and 5% CO2. Cells were examined after 2–3 days and noted to be spindle-shaped, consistent with MSC morphology. Media were replaced as needed, at least weekly, until 70–80% confluence, at which point the cells were de-adhered and passaged. Each batch of P-MSCs were characterized by function and phenotype. The former used the ability of P-MSCs to undergo multilineage differentiation towards adipocyte and osteocyte. Phenotypic studies used flow cytometry for specific MSC markers (see below).
P-MSC Differentiation
P-MSCs were differentiated with kits and followed manufacturer’s recommendations (Lonza, Basel, Switzerland) [12, 14]. The differentiated cells were fixed with formalin and stained with Oil Red O for adipogenic cells and silver nitrate for osteogenic cells. The images were captured with an inverted Nikon TMS microscope (100x).
Flow Cytometry
P-MSCs were subjected to direct labeling with fluorchrome-conjugated anti-CD29, -CD90, -CD105, and -CD45. The cells were analyzed on the FACS Caliber (Becton Dickinson, Franklin Lakes, NJ).
P-MSC Characterization
The P-MSCs were studied for function using multilineage differentiation towards adipocyte and osteocyte, as per manufacturer’s recommendations (Lonza, Basel, Switzerland) [12, 14]. Following differentiation, cells were fixed with formalin and stained with Oil Red O for adipogenic differentiation and silver nitrate for osteogenic differentiation. Nikon TMS microscope was used for image capture.
P-MSCs were analyzed for known markers by flow cytometry. This was achieved by labeling with fluorochrome-conjugated anti-CD29, -CD90, -CD105, and -CD45. The cells were analyzed on an FACS Caliber (Becton Dickinson, Franklin Lakes, NJ).
Isolation and Quantification of Exosomes
EVs were isolated from the media of P-MSCs by differential ultracentrifugation, as described [28]. Briefly, the media were serially centrifuged at 2000 g to remove residual cells and large debris, 10,000 g to clear large vesicles and debris, and 100,000 g to pellet the exosomes. The pellet was resuspended in PBS and centrifuged at 130,000 g to clear soluble contaminants. Exosomal extracts were analyzed by western blot for specific proteins. The total number of exosomes was determined by nanoparticle tracking analysis (NTA) using the NanoSight LM10 system [28].
P-MSC Treatment
P-MSCs from PE subjects were treated with EVs from healthy P-MSCs with 105–108 EV particles, or with ASA (1 mM freshly prepared). This dose of ASA was determined, based on previous studies [14]. ASA was added at time 0 and then readded at 24 h. EVs were incubated for 24 or 48 h and then used as third party cells in mixed lymphocyte reaction (MLR).
One Way Mixed Lymphocyte Reaction (MLR)
MLR was used as a model of inflammation to assess the capacity of the treated PE-derived P-MSC to be licensed into immune suppressor cells [12]. PE and healthy P-MSCs were treated with EVs, as indicated above, and used as third party cells in the one-way MLR. Parallel studies used untreated P-MSCs. The MLR was generated with peripheral blood mononuclear cells (PBMCs) from two healthy donors, as described [12, 16]. The PBMCs were isolated by Ficoll Hypaque density gradient, washed and then resuspended in DMEM with 10% FCS. One donor PBMCs were assigned to be the stimulator cells and were irradiated with 5000 rads with a Cesium (137Cs) source irradiator. The other donor’s PBMCs were assigned to be the responder cells. Cells from both donors were plated at equal concentrations and incubated at 37∘C and 5% CO2 for 72 h with and without the addition of P-MSC (3 104). At 72 h, the cells were pulsed with 1 Ci of tritiated thymidine. After 16 h, the cells were harvested on a Cambridge Technology PHD Cell Harvester. The filters were assessed for tritiated thymidine incorporation on a Beckman Coulter LS6500 Liquid Scintillation Counter.
Western Blot
Western blot was conducted with extracts from EV or P-MSCs, as described in [28]. Briefly, the samples were subjected to snap freeze with protease inhibitor and MPER. Total protein was determined using BioRad Bradford protein assay. Extracts (5–15 g) were electrophoresed on 10% SDS-PAGE and then transferred onto PVDF membranes. A Precision Plus Protein Kaleidoscope Ladder was used to assess molecular weights. Membranes were blocked with 2% milk for 20 mins. Primary antibodies were added at 4∘C overnight; HRP-conjugated secondary antibodies were added at 4∘C for 2 h. The membranes were imaged on a ChemiDoc XRS+ (BioRad). Membranes were stripped and re-probed for other proteins and -actin. Band densities were normalized to -actin using Un-Scan-It software (Silk Scientific Inc, Orem, Utah).
Statistical Analysis
Descriptive statistics, t-tests, one-way ANOVA with post hoc t-test with Bonferroni correction were performed in Prism GraphPad.
Results
P-MSC Characterization
P-MSCs were cultured from the chorionic membrane. We previously determined consistency with respect to ease of expanding P-MSCs from the chorionic membrane. P-MSCs were determined to be of fetal origin. This was done with P-MSCs from male neonates showing male-associated gene SRY, as previously shown [14]. The morphology of P-MSCs was spindle-shaped, which is consistent for P-MSCs, regardless of the presence of PE [14]. Functionally, P-MSC were induced to become adipogenic or osteogenic cells as previously described (Figure 1A) [14]. Flow cytometry indicated positive labeling for MSC markers, CD29, CD90, and CD105, and negative for the hematopoietic cell marker, CD45 (Figure 1B).
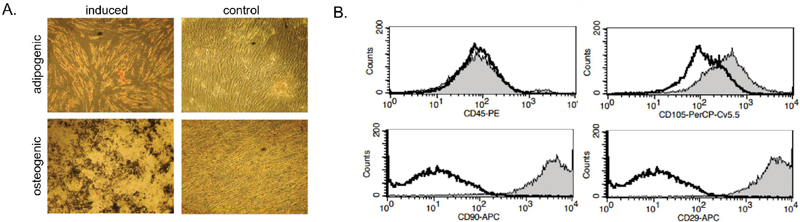
Figure 1 Characterization of P-MSCs. (A) Shown is a representative differentiation of P-MSCs towards adipogenic or osteogenic cells. (B) Representative histograms from flow cytometry results of P-MSCs labeled for the shown MSC markers.
Validating EV P-MSCs
We quantitated EVs from P-MSCs with a NanoSight. We also determined that the average size of the particles was 120 nm (Figure 2A). Western blot indicated that the EVs did not include cellular proteins as indicated by undetectable band for Histone as compared to bright bands in the cellular extract (Figure 2B). The EVs showed bands for exosomal protein, Alix (Figure 2A).
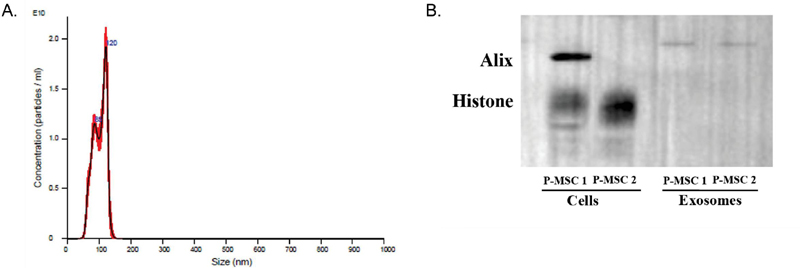
Figure 2 Quantification and analysis of EVs. (A) Use of nanoparticle tracking analysis (NTA) of extracellular vesicles was used to confirm exosomes by size and quantify the particles. (B) Western blot for Alix with protein extracts from P-MSCs and the released EVs. The membrane was blotted with histone.
Effects of EVs From Healthy P-MSCs to Restore the Licensing Function of PE P-MSC
In this set of studies we tested if EVs from healthy MSCs could restore the function of P-MSCs from PE subjects. We collected EVs from healthy P-MSCs and then added them to PE P-MSCs. After 48 h, we assessed the function of the treated PE P-MSCs. We placed the EV treated P-MSCs into a one-way MLR. If the function of PE P-MSCs is restored this should result in reduced proliferation of the MLR [12]. Since we previously showed restoration of PE P-MSCs by ASA, we included ASA stimulation as a control P-MSCs in the MLR.
We presented the proliferation of cells in MLR as stimulation index (S.I.). The S.I. was similar between the baseline MLR and untreated PE P-MSCs, indicating failed ability of the latter cells to suppress the proliferation within the MLR (Figure 3A). This contrasted with immune suppression by ASA treated P-MSCs, which showed significant () suppression in the MLR (Figure 3A), consistent with previous studies [16]. Next, we studied the effects of P-MSCs treated with EVs. The PE P-MSCs were treated with 105–108 EVs for 24 and 48 h. Both time course treatments and all doses of EVs showed significant () immune suppression as compared with untreated PE P-MSCs (Figures 3B and 3C). These studies indicated rapid and efficient restoration of PE P-MSCs when exposed to EVs from healthy P-MSCs.
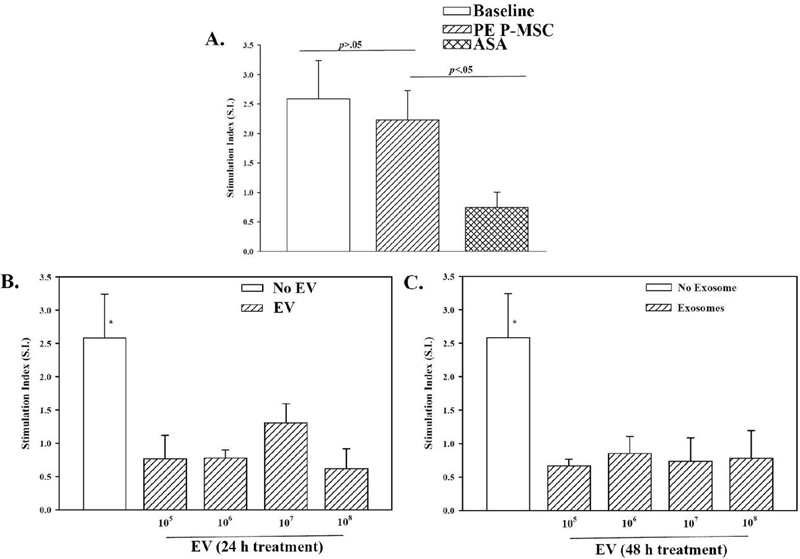
Figure 3 EVs from P-MSCs on the licensing of PE P-MSCs. (A) One-way MLR was performed with untreated P-MSCs or ASA treated P-MSCs. The responses were compared with studies without P-MSCs (positive control). The results are shown for three biological replicates. (B) P-MSCs were treated with healthy P-MSC-derived exosomes for 24 h (B) or 48 h (C) prior to addition of the PE P-MSCs as third-party cells.
EVs From Healthy P-MSCs Alter Cell Cycle Protein in PE-MSCs
We previously showed a decrease in cell cycle proteins when PE P-MSCs were treated with ASA [14]. Since healthy EVs mediated normal immune function as ASA (Figure 4), we asked if the EVs show evidence of decreased proliferation of PE P-MSCs. We exposed the PE P-MSC with 106 or 107 EVs for 24 and 48 h. The cell extracts were analyzed for p21 and CDK4. Although p21 was markedly decreased, it remained high, which is consistent with a decreased cell cycle as previously shown for ASA (Table 2) [14]. Since p21 inhibits CDK proteins, we noted reduced levels of CDK4 at all time points (Table 2) [29].
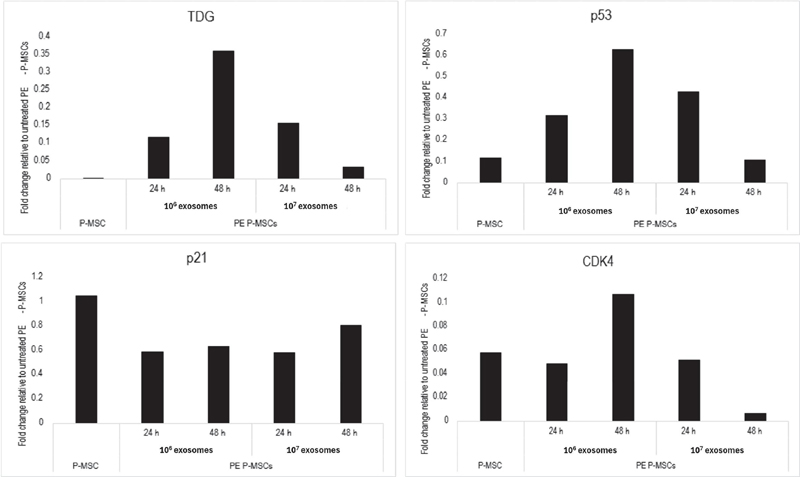
Figure 4 Exosome treatment alters PE P-MSC cell cycling. Cell cycle-associated proteins previously found to be altered in PE P-MSCs following aspirin treatment were measured followed PE P-MSC exposure to healthy exosomes for 24 or 48 h. Untreated healthy and PE P-MSCs were used as a control. Band density was normalized to -actin and reported as a fold change relative to untreated PE P-MSCs.
Increased level of p21 can be regulated by p53 [29]. We reported on increased p53 when PE P-MSCs were stimulated with ASA [14]. In this study, when P-MSCs were treated with EV we noted decreased p53 (Table 2). Since p53 is also involved in DNA repair and we previously reported increased in the basic excision repair protein, TDG, we examined its expression in the EV treated PE P-MSCs. In contrast to previous findings, we noted a decrease in TDG. The significance of these findings is discussed below.
Discussion
This study reports on the ability of EVs from healthy P-MSCs to restore the function of PE P-MSCs. Specifically, so they may be licensed immune suppressor cells (Figure 3). This effect was similar to that of low dose ASA (Figure 3) [16]. However, the mechanism by the data suggested that EVs might be important to sustain a stable restoration in PE P-MSCs, which is discussed in the next paragraph.
Table 2 Relative expression of cell cycling proteins
| Normalized Densities (% Difference from PE P-MSCs) | |||||
| Untreated | 106 EVs | 107 EVs | |||
| Cycling | PE | ||||
| Markers | P-MSCs | 24 h | 48 h | 24 h | 48 h |
| p21 | 0.93 | 0.55 (40.9%) | 0.60 (36.2%) | 0.54 (41.6%) | 0.76 (18.9%) |
| CDK4 | 1.86 | 0.09 (95.1%) | 0.20 (89.3%) | 0.10 (94.8%) | 0.01 (99.4%) |
| p53 | 0.39 | 0.12 (68.2%) | 0.25 (36.9%) | 0.17 (56.9%) | 0.04 (89.2%) |
| TDG | 0.31 | 0.04 (88.1%) | 0.11 (63.9%) | 0.05 (84.3%) | 0.01 (96.8%) |
| The band densities were normalized with the densities of -actin. | |||||
Consistent with previous studies showing cell cycle quiescence with ASA, we noted a decrease in CDK4 with EVs (Table 2) [14]. This premise is based on insights from western blot for cell cycle proteins and DNA repair proteins. This correlated with reduced p21 but with levels to maintain cycling quiescence (Table 2). Since p21 and p53 are involved in DNA repair and previous studies show ASA mediating increased p53, we analyzed the EV treated P-MSCs for p53. The results indicated that p53 was decreased with EV treatment (Table 2). We premised that the EV restoration returned the cells to homeostasis without the need for overt DNA repair. This type of return to homeostasis was not seen with ASA, suggesting a more stable effect with EVs. It is possible that EVs entering the cells initiated a reset for stable restoration, as noted in another system with aged hematopoietic cells [17]. In contrast, ASA is easily degraded and the low dose ASA used in the study to mimic patient treatment could not sustain the short-lived restoration. In fact, previous studies showed that the effect of ASA required readding the stimulus for a functional outcome [14]. ASA could maintain DNA repair with the basic excision repair mechanism. We showed that TDG was increased with ASA [15]. However, this protein was decreased with EVs, further suggesting a more stable restoration as compared to ASA.
This study is highly significant since PE is a common disease of pregnancy, affecting one in 25 women in the United States, and contributes to tens of thousands of maternal and hundreds of thousands of fetal deaths world-wide every year [30]. The cause of PE is poorly understood with a lack of post-delivery. The only current preventative medication is ASA. However, despite its use, the disease burden remains high. The findings of this study might begin to find other mechanisms to treat PE.
The in vitro study to treat PE P-MSCs with EVs from healthy P-MSCs could be extrapolated to an in vivo situation. Specifically, a system of tissue homeostasis in which healthy cells within a microenvironment, through intercellular communication, maintains functional balance. This might be occurring in what is deemed a healthy pregnancy. It is likely that this type of repair is occurring during pregnancy. The question is, why is PE “triggered” in a population of otherwise healthy women?
Future study will need to identify the EV cargo and to determine how their entry into the PE P-MSCs cause changes at the transcriptomic level and changes in epigenes. The in vitro studies were short term exposure to EVs. It would be interesting to conduct similar studies by readding EVs with an increased number of particles. These proposed studies would recapitulate the in vivo condition when the healthy EVs are continually released in the vicinity of the PE P-MSCs. Another major question is whether the EVs from PE P-MSCs can induce changes in the healthy EVs to facilitate the changes. The current studies used naïve EVs from healthy P-MSCs. Despite the positive functional changes, it would be important to use EVs from healthy P-MSCs that were exposed to secretome from PE P-MSCs.
EVs from healthy P-MSCs likely increase the proliferation of PE P-MSCs – a functional outcome shared with ASA treatment – enhancing the P-MSCs capacity to remain functional. This is one mechanism that might occur in vivo in an attempt to decrease PE-associated inflammation. It is likely that healthy P-MSCs – as well as maternal MSCs in the uterine cavity – release EVs to act on PE P-MSCs. However, in cases where PE manifests, the EVs might be insufficient for normal function, perhaps due to relatively few healthy P-MSCs. Future studies are needed to determine whether EVs derived from autologous maternal MSCs, either local to the placenta and uterine cavity (e.g. maternal P-MSCs from the decidua), or derived from other tissues (e.g. adipose, bone marrow) are capable of similarly enhancing PE P-MSC function. Similarly, coculture studies of maternal healthy MSCs with PE P-MSCs should be done to determine if these healthy maternal MSCs are capable of restoring PE P-MSCs, including what ratio of fetal:maternal MSCs may be needed. Given that MSCs from various tissues can exert slightly different functions based on their prior exposures, MSCs from specific tissues may prove, more or less, effective in this manner [14, 31]. This is an aspect of particular interest given that MSCs are known to experience cross-talk with other aberrant cells in their microenvironment, both to alter the function of the aberrant cells and whereby the aberrant cells alter the seemingly healthy MSC’s function [28]. Such studies will expand our understanding of the possibility of maternal autologous MSC transplant in treatment of PE.
In summary, EVs isolated from healthy P-MSCs may serve as an alternative or adjuvant treatment to ASA to mitigate development of, or possibly treat, preeclamptic P-MSCs to restore them towards a healthy phenotype, seen as an improved anti-inflammatory function. This improved anti-inflammatory function may reverse inflammatory damage from PE. Further studies are needed to assess how exosomes can reverse the PE phenotype in vivo; to understand if exosomes can be used instead of or as an adjunct to aspirin in the prevention of PE, and/or as a treatment once PE has already developed. As exosomes are immunologically inert, they could be used in vivo, including from autologous sources. However, understanding optimal timing, dosage, and route of administration are necessary. It is important to understand how quickly the exosomes are taken up and their ability to reach target tissues. Development of therapeutics that selectively promote healthy placental cell exosome secretion to stem the disease process may be the answer [11].
Acknowledgements
This work is in partial fulfillment of the fellowship of Jessica Greenberg.
Funding
This project was funded by a grant from the Metavivor Foundation.
Conflict of Interest
The authors have no conflicts of interest to report.
References
[1] Ford, N. D., S. Cox, J. Y. Ko, L. Ouyang, L. Romero, T. Colarusso, C. D. Ferre, C. D. Kroelinger, D. K. Hayes, and W. D. Barfield. 2022. Hypertensive Disorders in Pregnancy and Mortality at Delivery Hospitalization - United States, 2017-2019. MMWR Morb Mortal Wkly Rep 71: 585–591.
[2] Say, L., D. Chou, A. Gemmill, Ö. Tunçalp, A. B. Moller, J. Daniels, A. M. Gülmezoglu, M. Temmerman, and L. Alkema. 2014. Global causes of maternal death: a WHO systematic analysis. Lancet Glob Health 2: e323–e333.
[3] Hoyert, D. L., and A. M. Miniño. 2020. Maternal Mortality in the United States: Changes in Coding, Publication, and Data Release, 2018. Natl Vital Stat Rep 69: 1–18.
[4] Hutcheon, J. A., S. Lisonkova, and K. S. Joseph. 2011. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pract Res Clin Obstet Gynaecol 25: 391–403.
[5] 2020. Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222. Obstet Gynecol 135: e237–e260.
[6] Dekker, G. A., and B. M. Sibai. 1998. Etiology and pathogenesis of preeclampsia: Current concepts. Am J Obstet Gynecol 179: 1359–1375.
[7] Sargent, I. L., S. J. Germain, G. P. Sacks, S. Kumar, and C. W. G. Redman. 2003. Trophoblast deportation and the maternal inflammatory response in pre-eclampsia. J Reprod Immunol 59: 153–160.
[8] Rolnik, D. L., K. H. Nicolaides, and L. C. Poon. 2022. Prevention of preeclampsia with aspirin. Am J Obstet Gynecol 226: S1108–s1119.
[9] Henderson, J. T., K. K. Vesco, C. A. Senger, R. G. Thomas, and N. Redmond. 2021. Aspirin Use to Prevent Preeclampsia and Related Morbidity and Mortality: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. J Am Med Assoc 326: 1192–1206.
[10] 2021. ACOG Practice Advisory: Low-Dose Aspirin Use for the Prevention of Preeclampsia and Related Morbidity and Mortality.
[11] Shi, H., Z. Yang, J. Cui, H. Tao, R. Ma, and Y. Zhao. 2024. Mesenchymal stem cell-derived exosomes: a promising alternative in the therapy of preeclampsia. Stem Cell Res Ther 15: 30.
[12] Potian, J. A., H. Aviv, N. M. Ponzio, J. S. Harrison, and P. Rameshwar. 2003. Veto-like activity of mesenchymal stem cells: functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol 171: 3426–3434.
[13] Galipeau, J., M. Krampera, J. Barrett, F. Dazzi, R. J. Deans, J. DeBruijn, M. Dominici, W. E. Fibbe, A. P. Gee, J. M. Gimble, P. Hematti, M. B. Koh, K. LeBlanc, I. Martin, I. K. McNiece, M. Mendicino, S. Oh, L. Ortiz, D. G. Phinney, V. Planat, Y. Shi, D. F. Stroncek, S. Viswanathan, D. J. Weiss, and L. Sensebe. 2016. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 18: 151–159.
[14] Romagano, M. P., L. S. Sherman, B. Shadpoor, M. El-Far, S. Souayah, S. H. Pamarthi, J. Kra, A. Hood-Nehra, J. P. Etchegaray, S. F. Williams, and P. Rameshwar. 2022. Aspirin-Mediated Reset of Preeclamptic Placental Stem Cell Transcriptome - Implication for Stabilized Placental Function. Stem Cell Rev Rep 18: 3066–3082.
[15] Krishnamoorthy, K., L. S. Sherman, M. P. Romagano, M. El Far, J. P. Etchegaray, S. F. Williams, and P. Rameshwar. 2023. Low dose acetyl salicylic acid (LDA) mediates epigenetic changes in preeclampsia placental mesenchymal stem cells similar to cells from healthy pregnancy. Placenta 137: 49–58.
[16] Powell, K. A., L. S. Sherman, B. Shadpoor, S. F. Williams, and P. Rameshwar. 2024. Restore Veto Property in Low Dose Aspirin/ASA Treated Preeclampsia Placenta Mesenchymal Stem Cells: Insights Into ASA-mediated Clinical Response*. Intl J Transl Sci 2024: 133–152.
[17] Greco, S. J., S. Ayer, K. Guiro, G. Sinha, R. J. Donnelly, M. H. El-Far, L. S. Sherman, Y. Kenfack, S. H. Pamarthi, M. Gergues, O. A. Sandiford, M. J. Schonning, J. P. Etchegaray, and P. Rameshwar. 2021. Restoration of aged hematopoietic cells by their young counterparts through instructive microvesicles release. Aging (Albany NY) 13: 23981–24016.
[18] Chu, Y., W. Chen, W. Peng, Y. Liu, L. Xu, J. Zuo, J. Zhou, Y. Zhang, N. Zhang, J. Li, L. Liu, K. Yao, G. Gao, X. Wang, R. Han, C. Liu, Y. Li, H. Zhou, Y. Huang, and Y. Ye. 2020. Amnion-Derived Mesenchymal Stem Cell Exosomes-Mediated Autophagy Promotes the Survival of Trophoblasts Under Hypoxia Through mTOR Pathway by the Downregulation of EZH2. Front Cell Dev Biol 8: 545852.
[19] Liu, H., F. Wang, Y. Zhang, Y. Xing, and Q. Wang. 2020. Exosomal microRNA-139-5p from mesenchymal stem cells accelerates trophoblast cell invasion and migration by motivation of the ERK/MMP-2 pathway via downregulation of protein tyrosine phosphatase. J Obstet Gynaecol Res 46: 2561–2572.
[20] Chen, Y., H. Ding, M. Wei, W. Zha, S. Guan, N. Liu, Y. Li, Y. Tan, Y. Wang, and F. Wu. 2020. MSC-Secreted Exosomal H19 Promotes Trophoblast Cell Invasion and Migration by Downregulating let-7b and Upregulating FOXO1. Mol Ther Nucleic Acids 19: 1237–1249.
[21] Yang, Z., N. Shan, Q. Deng, Y. Wang, Y. Hou, J. Mei, and Z. Wu. 2021. Extracellular vesicle-derived microRNA-18b ameliorates preeclampsia by enhancing trophoblast proliferation and migration via Notch2/TIM3/mTORC1 axis. J Cell Mol Med 25: 4583–4595.
[22] Cui, J., X. Chen, S. Lin, L. Li, J. Fan, H. Hou, and P. Li. 2020. MiR-101-containing extracellular vesicles bind to BRD4 and enhance proliferation and migration of trophoblasts in preeclampsia. Stem Cell Res Ther 11: 231.
[23] Jiang, Y., T. Luo, Q. Xia, J. Tian, and J. Yang. 2022. microRNA-140-5p from human umbilical cord mesenchymal stem cells-released exosomes suppresses preeclampsia development. Funct Integr Genomics 22: 813–824.
[24] Yu, H., Y. Pan, M. Dai, X. Wang, and H. Chen. 2023. Mesenchymal Stem Cell-Originated Exosomal Lnc A2M-AS1 Alleviates Hypoxia/Reperfusion-Induced Apoptosis and Oxidative Stress in Cardiomyocytes. Cardiovasc Drugs Ther 37: 891–904.
[25] Cao, L., H. Xu, G. Wang, M. Liu, D. Tian, and Z. Yuan. 2019. Extracellular vesicles derived from bone marrow mesenchymal stem cells attenuate dextran sodium sulfate-induced ulcerative colitis by promoting M2 macrophage polarization. Int Immunopharmacol 72: 264–274.
[26] Shahir, M., S. Mahmoud Hashemi, A. Asadirad, M. Varahram, M. Kazempour-Dizaji, G. Folkerts, J. Garssen, I. Adcock, and E. Mortaz. 2020. Effect of mesenchymal stem cell-derived exosomes on the induction of mouse tolerogenic dendritic cells. J Cell Physiol 235: 7043–7055.
[27] Xin, L., X. Lin, F. Zhou, C. Li, X. Wang, H. Yu, Y. Pan, H. Fei, L. Ma, and S. Zhang. 2020. A scaffold laden with mesenchymal stem cell-derived exosomes for promoting endometrium regeneration and fertility restoration through macrophage immunomodulation. Acta Biomater 113: 252–266.
[28] Sandiford, O. A., R. J. Donnelly, M. H. El-Far, L. M. Burgmeyer, G. Sinha, S. H. Pamarthi, L. S. Sherman, A. I. Ferrer, D. E. DeVore, S. A. Patel, Y. Naaldijk, S. Alonso, P. Barak, M. Bryan, N. M. Ponzio, R. Narayanan, J.-P. Etchegaray, R. Kumar, and P. Rameshwar. 2021. Mesenchymal Stem Cell–Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res 81: 1567–1582.
[29] Karimian, A., Y. Ahmadi, and B. Yousefi. 2016. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 42: 63–71.
[30] Dawson, E. L., Khoury, Muin J. 2022. Preeclampsia, Genomics and Public Health.
[31] Sherman, L. S., M. Shaker, V. Mariotti, and P. Rameshwar. 2017. Mesenchymal stromal/stem cells in drug therapy: New perspective. Cytotherapy 19: 19–27.
International Journal of Translational Science, Vol. 2, 113–130
doi: 10.13052/ijts2246-8765.2025.021
© 2026 River Publishers