A Novel Customized Big Data Analytics Framework for Drug Discovery
A. Jainul Fathima1,* and G. Murugaboopathi2
1Research Scholar
2Associate Professor
1,2Department of Computer Science and Engineering, Kalasalingam Academy of Research and Education, Krishnankoil, Tamilnadu, India
E-mail: jainulfathima.a@klu.ac.in
*Corresponding Author
Received 03 January 2018; Accepted 03 April 2018;
Publication 28 April 2018
Abstract
Drug discovery is related to analytics as the method requires a technique to handle the extremely large volume of structured and unstructured biomedical data of multi-dimensional and complexity from pharmaceutical companies. To tackle the complexity of data and to get better insight into the data, big data analytics can be used to integrate the massive amount of pharmaceutical data and computational tools in an analytic framework. This paper presents an overview of big data analytics in the field of drug discovery and outlines an analytic framework which can be applied to computational drug discovery process and briefly discuss the challenges. Hence, big data analytics may contribute to better drug discovery.
Keywords
- Big Data
- Analytics
- Framework
- Drug Discovery
1 Introduction
The Pharmaceutical industry is seeing a growth in data from multiple sources that include Research and Development (R&D) process, patient’s clinical trial record and from healthcare professionals. Reports say the expense of developing a new drug is approximate $2.6 billion and it will rise with a growth rate of 8.5 percent per annum [1]. The R&D data generated from multiple sources require effective processing and integration using powerful analytical and visualizationtools. Such data when efficiently used will help to better identify new potential drug candidate and develop into effective approved medicines more quickly. All companies are forced to use advanced analytical methods and the way in which this wealth of data is used is getting more and more refined with time [2]. In drug discovery, the pharmaceutical industry spends a vast amount of money in screening compounds to test in preclinical trials. To speed up the process, drug companies are using predictive models to search massive virtual databases of molecular and clinical data. With the knowledge of chemical structure, diseases/targets and other characteristics, the data analysts look for the likely drug candidates.
In the era of Big Data in drug development, High-Throughput Screening Techniques (HTS) like Next-Generation Sequencing (NGS) are helping the researchers to analyze patients in terms of genome and proteome [3]. Initiatives are taken for precision medicine for disease treatment considering the changes in the molecular system. This technology-driven approach can develop success rate in drug development process with less expense and improve patient health with direct involvement. With the advancement in computing techniques data generation, storage and processing cost are reduced. To meet the modern research needs, there is a need for analytic tools to analyze and interpret terabytes of data. Traditional mathematical computational tools are not armed to handle these Big Data sets due to the problems resulting from multi-dimensional and association in genetic data. With the advancement in Big Data and technology, merging of analytic methods and biological systems can be used to develop flexible frameworks to discover lead molecule for the future of drug development.
To enable a data-driven drug development method and to use NGS data; data mining and machine learning techniques are essential. It is recommended to use implementation tools that control the power of multiple algorithms and to combine multiple tools in the same working environment [4]. Informatics has to be used for speedy and efficient work in higher learning in all branches of knowledge when informatics is used in the study of biology-related areas we usually referred to as bioinformatics. It has been an integral tool for learning at a higher level. Its application is inevitable and necessary for meaningful research activity. In this study, we are utilizing it for understanding and development of diseases and drug development. The bioinformatics tools are necessary for the efficient and accelerated discovery of drugs. Thereis a large number of bioinformatics tools available for various stages of drug discovery process starting from Homology modeling of the target protein to docking of ligand to the protein. A large number of virtual screening databases are also available. A survey on Computational approaches [5] and implementation [10] of drug discovery is reported gives knowledge about various tools available for computational drug discovery process.
This article provides an overview of bigdata analytics for drug discovery. First, we discussed the big data in a pharmaceutical industry that can be considered for drug discovery application. Second, we summarize the application of data mining, machine learning algorithms to handle the different varieties of data. Third, we propose an idea of integrating big data and tools into an analytic framework for drug discovery. Lastly, we identified some challenges in implementing and offer a conclusion and future research direction in this field.
1.1 Pharmaceutical Big Data Sources for Drug Discovery
Drug Discovery is a multi-step process of developing drugs for effective treatment of a disease. The process starts from searching for ligand libraries to find the lead compound, evaluating and optimizing the lead compound with biological activity against the specific target. A Target or Receptor is a macromolecule or protein structure where the ligand binds. The main aim of applying analytics is to ease the computational drug discovery process [6]. There area large number of protein databases which stores biological information for analysis and drug discovery application. Here we reported some of the protein databases. Protein databases contain more than 300,000 protein sequences. Some of the databases include SWISS-PROT, Protein information resource (PIR), TrEMBL, GenPept, RefSeq, Protein Data Bank (PDB) [7]. The PDB contains the 3D structure of macromolecules determined by X-Ray crystallography and NMR techniques. The major challenge involved is to manage and integrate the existing biological databases for improving the drug discovery process. Most of the drugs available in the market came from a screening of natural compound ligand libraries. This kind of ligand libraries is constructed using computational tools. Virtual HTS is used to filter large chemical databases to find the lead compound. By using the parallel processing clusters, we can filter upto 100,000 ligands per day. To perform virtual screening process, a virtual library is generated. Several ligand databases are available that gives all the information about known chemical compounds. Some of the ligand databases include PubChem, drug bank, chemDB, ZINC, LIGAND [8]. Ligand databases are often built with better quality ligands which satisfy drug-likeness properties. Drug-likeness is commonly evaluated using Lipinski rule of 5 [29]. Relibase is a protein-ligand database which contains PDB structures with a ligand binding interaction. Various other data taken from the pharmaceutical industry for this application include repurposing of drugs data, data related to clinical trials. Transforming the publically available databases to knowledge is the major goal of bioinformatics application. Transforming these heterogeneous data to a form for extracting the hidden knowledge is dependent on data preparation, cleaning and integrating which is possible by applying big data transformation methods like datamining and machine learning methods.
2 Related Works
2.1 Data Mining Techniques for Drug Discovery
The main goal of data mining is to extract information from a large amount of biological data. Effective data mining became critical to drug development. The process starts from identification of valid target which involves isolation of diseased tissue and analyzing its sequence and gene expression. After finding the diseased gene it is important to validate them as targets. The high Computational complexity involved in sequence alignment which can be solved using dynamic programming [9]. It is a simple and fast method. The disadvantage of this approach is any mistake made from intermediate alignment cannot be corrected. Another approach involves using a probabilistic technique for multiple alignments. Validation of target verifies the DNA that is involved in a disease process. This is the crucial step. Target validation can be done using DNA microarray technology. The disadvantage of this approach is when the data set contains more than five classes may not produce accurate results. Hence, there is a need for developing big data analytic algorithms for analyzing multiple class expression data. To make use of the data from these databases require software tools to extract data, compare the sequences, pattern analysis and for visualization. The most useful data mining techniques are an association, Classification, Sequence path analysis, Clustering, and forecasting. Each ligand consists of multiple attributes. The interaction between the attributes could be difficult to find. In mining concept, Classification is used to understand the relationship between various conditions and features. For case, there can be a training data set with two classes of ligands, Set of all ligands and ligand that binds to the protein. It is important to classify the ligands so that new ligand can be obtained. Major classification methods include decision tree, neural networks, Support Vector Machine (SVM). The concept of SVM is considered to be accurate for drug discovery application. When there is less number of attributes the decision tree classifier works better than SVM. In the decision tree concept, the internal node is categorized with a set of ranges. A range is linked with a path to a leaf node. If the particular attribute of the ligand falls in the range, then the search travels down the tree through the corresponding path. Since for drug discovery process, the important criteria is to deal with high dimensional data, so decision tree concept will not be applicable to drug discovery concept. Drug Likeness prediction can be predicted using SVM. The related research reported in the literature for the application of these data mining concept for drug discovery process are discussed.
Wencong, Lu [11] briefly introduces data mining concept with a focus on application in chemistry to understand the activity relationship of a drug. Also in this paper, the author worked on SVM and pattern recognition techniques to mine the chemical compound library to achieve industrial optimization. L.M. Shi [12] proposed an integrated approach based on data mining concept. This paper highlights some of the existing statistical, machine learning and visualization techniques which can be applied to drug discovery using data mining concept as a base. Interestingly, David J. Wild [13] reviewed about various publically available data set related to drug discovery process and discussed the application of data mining approach and suggested applying advanced analytical methods to handle larger heterogeneous data sets for drug discovery. Bryant [14] discuss target identification and lead optimization using data mining tools which are the most needed requirement of drug discovery process and it should be a cost-effective and reliable method for drug discovery process. Yongliang Yang reviewed the application of datamining concept for drug discovery process [15]. Most of the data mining concept use statistical methods and it is termed as machine learning methods.
2.2 Machine Learning Algorithms for Drug Discovery
Machine learning programs enable the computer to learn from experiences and adapt their behavior. The existing bioinformatic research area such as Gene finding, Protein folding prediction, Pattern identification uses machine learning approaches to discover knowledge to analyze and predict disease and to find associations in biological data. Some of the most popular approaches include neural networks, genetic algorithms. In the drug discovery process, computational docking is the mechanism to predict how the small molecule (ligand) binds to the protein target so as to form a stable molecule. Many binding poses are calculated and ranked using the scoring function. Scoring function encompasses the predictive mathematical model which calculates a score called binding free energy. The effective scoring function can produce appropriate drug molecule. Hence Machine learning techniques can be applied to design a highly accurate scoring function. Zhang, L., [16] propose a technique to apply machine learning algorithm to predict the scoring function and suggested the idea to use deep learning algorithm in this study. Deep learning algorithms are used for pattern recognition speech recognization and natural language processing. This concept represents data in multiple layers. Hecht, David [17] is a research overview article which gives an idea to adapt the machine learning and computational intelligence tools in the field of drug discovery and development. A step by step procedures to build machine learning based classification and regression models using open source tools which are used to build reliable and predictive models in chemoinformatics is reported [18]. The scientist also suggests that machine learning technique are suitable for high dimensional data and models to build can be used for prediction and that can be applied at various stages of drug discovery including virtual screening, molecular docking, and target prediction [19]. Various machine learning tools like SVM, Genetic programming, particle swarm optimization are used in pharmaceutical research and development [20]. Due to the advancement in high throughput screening which results in the generation of billions of molecules. This terabyte of data of high velocity and veracity led to consider drug discovery as an Analytic problem. The Next section outlines the idea of integrating previously explained big data sources and big data transformation into an analytic framework.
3 Bigdata Analytic Framework for Drug Discovery
Big data analytics tools are difficult, programming specific and require the application of different varieties of skills. Big data collected from the pharmaceutical companies will be of structured, unstructured and semi-structured data sources often will be in various formats such as .pdb, .mol, .csv, text, etc., For the need of drug discovery, these data has to be extracted and converted into raw data and stored in a data warehouse which contains data aggregated and made ready for processing. Various drug discovery applications like high throughput screening, virtual screening, docking studies, Toxicity prediction, and Lead optimization technique can be performed with these data. Drug discovery is also an area that increasingly produces more data in many forms. Therefore, drug discovery process is likely to get benefit from new forms of big data. The researchers make decisions about treatment options with the data related to specific illness. Data elements are stored and managed by the various organizations. Various clinical researches can be used to determine the effectiveness of a drug. To handle increasing data size, dimensionality and complexity of data we need an analytic framework to overcome the drawback of the traditional data processing system [21].
The standard solution for implementing analytics for drug discovery includes R, Matlab, Hadoop and various analytic tools [22]. Due to the rapid growth of data and unclear endpoints we need a system that should perform analytics exactly like R but should work for Tetra Byte (TB) size data sets which has multiple users like analysts, developers, and scientist. The solution to this is the development of data analytic framework. The major requirement for this framework is it should be available as open source and the data model should be nested with multi-dimensional arrays and should support multi-dimensional storage. When the data model is the multidimensional array it is easy to know which compounds have a similar transcriptional profile and what proteins bind to the lead compound and it can be extended by correlation and multidimensional statistics. It should also incorporate uncertainty and data persistence. Considering the requirements and analyzing the various big data analytic tools for drug discovery there is in need to blend new technologies with an existing system.
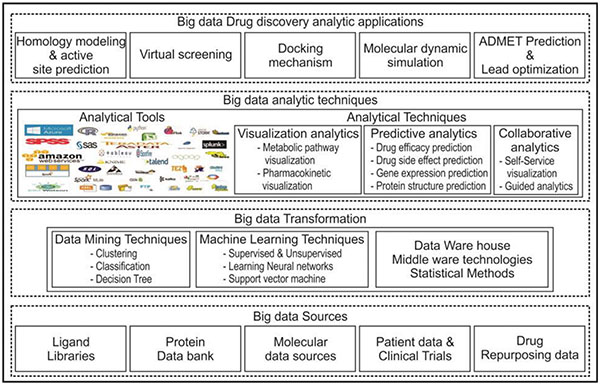
Figure 1 Big data Analytic framework for drug discovery.
Figure 1. Proposes an analytic framework that is specifically for drug discovery process. This analytic framework integrates publicly available big data sources, data mining tools, visualization and analytic tools. This has four section while looking at it from bottom to top of the framework. The big data sources which include how the data is collected, accessed, ingested, and organized. The various big data sources for drug discovery are explained in the previous section. The collected data has to be preprocessed using big data transformation by applying datamining and machine learning concepts and stored in a data warehouse. The transformed data can be applied with analytical tools. Various types of analytics can be done that include predictive analytics [23], visualization analytics [24], and collaborative analytical approach [25]. Predictive analytics which includes drug efficacy prediction, drug side effect prediction, Gene expression prediction, protein folding prediction, protein structure prediction. Next set of analytics is visual analytics for metabolic pathway visualization and pharmacokinetic visualization. Finally, for collaborative analytics, the working environment is linked to real-time data.
Figure 2 Presents an outline methodology for drug discovery to implement an application like active site prediction, docking, molecular dynamic simulation, toxicity prediction and lead optimization. The methodology involves series of steps followed for drug discovery application. For example, consider a problem of finding a lead compound for a particular protein target. Initially, the protein structure is retrieved from the PDB and it is preprocessed. Various lead identification techniques like virtual screening, HTS techniques are applied to the ligand databases to find the lead compound. The binding affinity of protein-ligand interaction can be compared with the big data sources. The identified lead compound can be further optimized by applying combinatorial chemistry. Computationally performing these task with the support of big data analytic concept will make the drug discovery approach more beneficial in terms of cost and time. Some of the interesting applications of analytic platforms reported in the literature that includes Octopus [26] tool perform Virtual HTS by integrating docking and visualization tools. Scaffold Hunter [27] is a visual analytic framework for drug discovery. A major challenge in applying analytic concept is it needs high programming skills to manage various types of big data. In the next section, we have integrated virtual screening tool Autodock, visualization tool pymol, drug toxicity prediction tool within an analytic framework. We implemented in a platform for drug toxicity prediction based on machine learning algorithm. The database used here for analysis is obtained as a result of virtual screening the ligand database. Though there are many virtual screening tools available. Here, we have used Autodock tool [28] which is very fast in execution and provides high-quality predictions of ligand conformations and Auto Dock is free software and version 4 is distributed under the General Public License. The result of virtual screening is stored in the database for further analysis.

Figure 2 Outline methodology for drug discovery.
3.1 Integrating the Data Sources
In the big data analytical research, to get more benefit from the data, it needs to be integrated with the working process. The researchers can analyze and make a decision considering many factors. There are two categories of integration they are the integration of multiple big data sources in big data environment and the integration of unstructured big data sources with structured data. Generally, for traditional data processing approach data mining concepts are used for data preprocessing techniques but for big data environment, there is a need to combine tools that support batch integration same like Extract, Transform, Load (ETL) option with real-time integration across multiple sources. The pharmaceutical company needs to combine its data stored with the publicly available big data sources. The company will use the stored data to collect, aggregate, consolidate and deliver reliable data across the big data sources. For accomplishing the data integration in big data environment the data elements have to be mapped with common definitions and it is forward to operational data and data storage. A set of data services are developed to qualify the data to make it consistent. Further, to take a decision based on the results, the system should update the results periodically. The data integration process should be consistent and reliable. As an experimental study, the above-discussed framework and methodology are implemented for predicting the drug toxicity based on ADMET properties and prediction of accuracy is done based on machine learning algorithm. To implement this approach data preprocessing is done to remove the redundant and irrelevant data followed by feature extraction step which means extracting some certain characteristic attributes and generating a set of meaningful descriptors from the dataset. Based on the feature extraction result, the feature set is determined which distinguish the sample. Finally, classification is used to classify the data into two classes toxic and nontoxic based on the selected feature. For implementing the classification algorithm, here Naïve Bayes classifier is used. The performance is calculated based on the accuracy parameter.
3.2 Experiment and Results
As a case study, the integrated platform is used for lead identification followed by drug toxicity prediction. This platform integrates data warehouse, Auto Dock tool, Pymol, Toxicity prediction tool and Machine learning algorithms for further prediction. For performing the lead identification, there are series of steps to be followed and that includes database search, protein structure prediction, virtual screening and molecular docking and denova design. To perform docking and virtual screening studies, the integrated Autodock tool option is called as a function. The result of the virtual screening tool is a .csv file which can be visualized in the same platform. Further analysis can be done with the visual analytics tool integrated with the platform. Figure 3 shows the analytic platform integrated with a various tool for drug toxicity prediction. The database stored here is a structured .csv file which is generated by virtual screening the ZINC database to find the protein-ligand interaction which contains the details of 40,000 chemical compounds.
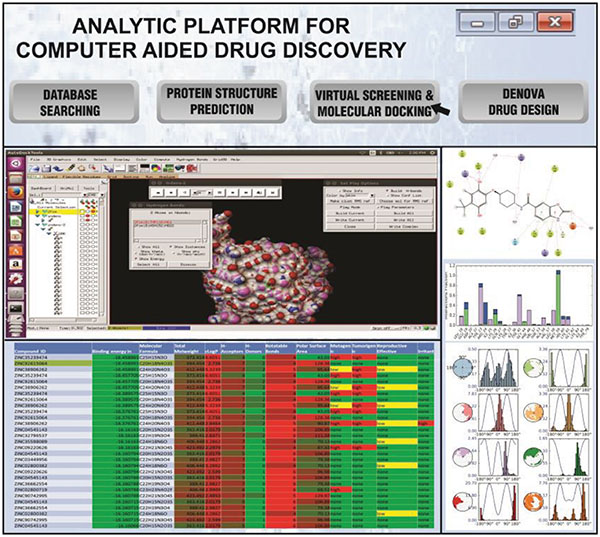
Figure 3 Integrated platform for drug toxicity prediction.
The dataset has the following attributes that include compound id, Binding energy, Total molecular weight, CLogP, H-Acceptor, H-Donar, Rotatable bonds, Polar Surface Area, Mutagenic, Tumorogenic, Reproductive Effect, Irritant. The values of the attributes are checked with the Lipinski rule of 5 [29] and classified into toxic and non-toxic classes. Each case of the dataset has one of two possible classes. toxic indexed with 0 or non-toxic indexed with 1. The class distribution for toxic 27360 (68.4%) and non-toxic 12640 (31.6%). The example view of data set is shown in Figure 3. The software tool for implementing this study is written in python and uses naïve Bayes classifier. Naïve Bayes classification follows supervised learning approach and it is an effective method for classification. The effectiveness of the algorithm is that to make a prediction it will consider the probability of each attribute belonging to each class.
Table 1 Results of Naïve Bayes classifier performance
| Number of Attributes | Feature Set | Accuracy |
| 12 | Compound id, Binding energy, Total molecular weight, CLogP, H-Acceptor, H-Donar, Rotatable bonds, Polar Surface Area, Mutagenic, Tumorogenic, Reproductive Effect, Irritant | 93.876 % |
This Naïve Bayes classifier considers the class prior probability and predictor prior probability to calculate the posterior probability and the probability of a specified outcome. The steps followed for implementing the naïve Bayes classifier using python is reported [30]. The attained accuracy obtained by using naïve Bayes classifier is 0.9387651009. The Formula that calculate the accuracy is Accuracy = [(TP + TN)/(TP + TN + FP + FN)] where TP (true positive), FN (false negative), FP (false positive), TN (true negative).
4 Challenges
The big data analytics platform for drug discovery must support certain functions to process different kinds of data. The factors to be considered when evaluating the analytic platform are availability, scalability, ability to manipulate at various levels, usability [31]. To be more effective, big data analytics for drug discovery should integrate most of the computer-aided drug design tools and it should be menu-driven and transparent to use.
5 Conclusion
The pharmaceutical companies can effectively use big data analytic techniques to find the lead molecule to develop into accepted and active drugs. This paper discusses various analytical approaches which can be appropriately applied at various stages of drug discovery approach. It is suggested that some of these above-mentioned methods can be more efficient. Researchers can pick choose the methodology enumerated above depending on which method suits the discovery process. Multiple approaches or combination of several approaches can make research work even more efficient to confirm research obtained by one approach. The framework will act as a guideline for the future research efforts in the area of big data analytics in drug discovery application. In future, the implementation of these advanced analytical methods which are technology-enabled can improve the success rate of drug discovery process. As the big data analytical techniques become more important, other issues such as privacy and security of biomedical data, establishing a standard and continually improving bioinformatics tools and databases would gather consideration.
References
[1] DiMasi, J. A., Feldman, L., Seckler, A.,and Wilson, A. (2010). Trends in Risks Associated With New Drug Development: Success Rates for Investigational Drugs. Clinical Pharmacology & Therapeutics. 87, 272–277.
[2] Yousefi, N., Mehralian, G., Rasekh, H. R., and Yousefi, M. (2017). New Product Development in the Pharmaceutical Industry: Evidence from a generic market. Iranian Journal of Pharmaceutical Research?: IJPR. 16(2), 834–846.
[3] Schmidt, Bertil, and Andreas Hildebrandt. (2017). Next-Generation Sequencing: Big Data Meets High-Performance Computing. Drug Discovery Today. 22, 712–717.
[4] Lusher, S. J., Mcguire, R., Schaik, R. C., Nicholson, C. D., and Vlieg, J. D. (2014). Data-driven medicinal chemistry in the era of big data. Drug Discovery Today. 19(7), 859–868.
[5] Fathima, A.J., Murugaboopathi, G., and Selvam, P. (2017). Computational Approaches in Drug Discovery: An Overview. International Journal of Advanced Research in Science and Engineering. 6(7), 189–195.
[6] Lusher, S. J., Mcguire, R., Schaik, R. C., Nicholson, C. D., and Vlieg, J. D. (2014). Data-driven medicinal chemistry in the era of big data. Drug Discovery Today, 19(7), 859–868.
[7] Hung, C., and Chen, C. (2014). Computational Approaches for Drug Discovery. Drug Development Research. 75(6), 412–418.
[8] Fathima, A., Murugaboopathi, G., and Selvam, P. (2018). Pharmacophore Mapping of ligand-based virtual screening, molecular docking and molecular dynamics simulation studies for finding potent NS2B/NS3 Protease Inhibitors as potential anti-dengue drug compounds. Current Bioinformatics, 13, doi:10.2174/1574893613666180118105659
[9] Babaie-Kafaki, S. (2016). Computational Approaches to Large-Scale Unconstrained Optimization. Studies in Big Data Big Data Optimization: Recent Developments and Challenges. 18, 391–417.
[10] Berman, H., Nakamura, H. and Henrick, K. (2005). The Protein Data Bank (PDB) and the Worldwide PDB. In Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics eds L. B. Jorde, P. F. Little, M. J. Dunn and S. Subramaniam. doi:10.1002/047001153X.g406303
[11] Wencong L. (2010). Data Mining and Discovery of Chemical Knowledge. In: Gaber M. eds Scientific Data Mining and Knowledge Discovery. Springer, Berlin, Heidelberg. p. 269–317.
[12] Shi L.M., Tong W.D. (2003). Data Mining: An Integrated Approach for Drug Discovery. In: Xing WL., Cheng J. eds Biochips. Biological and Medical Physics Series. Springer, Berlin, Heidelberg. p. 71–89.
[13] Wild, D. J. 2009. Mining large heterogeneous data sets in drug discovery. Expert Opinion on Drug Discovery. 4(10), 995–1004.
[14] Bryant, S. D. and Langer, T. (2013). Data Mining Using Ligand Profiling and Target Fishing. In Data Mining in Drug Discovery eds R. D. Hoffmann, A. Gohier and P. Pospisil. doi:10.1002/9783527655984.ch11
[15] Yongliang Yang, S. James Adelstein, Amin I. Kassis. (2009). Target discovery from data mining approaches, Drug Discovery Today, 14(3), 147–154.
[16] Zhang, L., Tan, J., Han, D., and Zhu, H. (2017). From machine learning to deep learning: Progress in machine intelligence for rational drug discovery. Drug Discovery Today, 22(11), 1680–1685.
[17] Hecht, David. (2010). Applications of machine learning and computational intelligence to drug discovery and development. Drug Development Research. 72(1), 53–65.
[18] Karthikeyan M., Vyas R. (2014). Machine Learning Methods in Chemoinformatics for Drug Discovery. In: Practical Chemoinformatics. Springer, New Delhi. P. 133–194.
[19] Jorissen, R. N., and Gilson, M. K. (2005). Virtual Screening of Molecular Databases Using a Support Vector Machine. J. Chem. Inf. Model. 45(3), 549–561.
[20] Barrett S.J., Langdon W.B. (2006). Advances in the Application of Machine Learning Techniques in Drug Discovery, Design, and Development. In: Tiwari A., Roy R., Knowles J., Avineri E., Dahal K. (eds) Applications of Soft Computing. Advances in Intelligent and Soft Computing, vol. 36. Springer, Berlin, Heidelberg.
[21] Raghupathi, W., and Raghupathi, V. (2014). Big data analytics in healthcare: Promise and potential. Health Information Science and Systems, 2, 3. doi:10.1186/2047-2501-2-3
[22] Oussous, A., Benjelloun, F., Lahcen, A. A., and Belfkih, S. (2017). Big Data technologies: A survey. Journal of King Saud University – Computer and Information Sciences. doi:10.1016/j.jksuci.2017.06.001
[23] Watson, C. (2004). New techniques and strategies in predictive ADME–Tox. Drug Discovery Today: BIOSILICO. 2(2), 55–56.
[24] Roberts, B. R. (2000). Screening informatics: Adding value with meta-data structures and visualization tools. Drug Discovery Today, 5, 10–14.
[25] Minna Allarakhia (2018) Evolving models of collaborative drug discovery: managing intellectual capital assets, Expert Opinion on Drug Discovery, doi: 10.1080/17460441.2018.1455659
[26] Maia, E. H., Campos, V. A., Santos, B. D., Costa, M. S., Lima, I. G., Greco, S. J., and Taranto, A. G. (2017). Octopus: A platform for the virtual high-throughput screening of a pool of compounds against a set of molecular targets. Journal of Molecular Modeling, 23(1), 26. doi:10.1007/s00894-016-3184-9
[27] Klein K., Kriege N., Mutzel P. (2013) Scaffold Hunter: Facilitating Drug Discovery by Visual Analysis of Chemical Space. In: Csurka G., Kraus M., Laramee R.S., Richard P., Braz J. (eds) Computer Vision, Imaging and Computer Graphics. Theory and Application. Communications in Computer and Information Science, vol. 359. Springer, Berlin, Heidelberg p. 176–192.
[28] Baba, N., and Akaho, E. 2011. VSDK: Virtual screening of small molecules using AutoDockVina on Windows platform. Bioinformation, 6(10), 387–388.
[29] Al-Lazikani, B. (2004). Rule of Five (Lipinski Rule of Five). Dictionary of Bioinformatics and Computational Biology. doi:10.1002/97804716501 26.dob1075
[30] Sun, H. 2005. A Naive Bayes Classifier for Prediction of Multidrug Resistance Reversal Activity on the Basis of Atom Typing. Journal of Medicinal Chemistry, 48(12), 4031–4039.
[31] Singh, D., and Reddy, C. K. (2014). A survey on platforms for big data analytics. Journal of Big Data, 2(1). doi:10.1186/s40537-014-0008-6
Biographies

A. Jainul Fathima received her B.Tech. degree in Information Technology from Anna University – Chennai in 2007 and M.Tech degree in Computer Science and Engineering from Anna University – Tirunelveli in 2009. She has 3 years of teaching experience. She is currently pursuing Ph.D. degree in Kalasalingam Academy of Research and Education, Krishnankoil. Her Research area includes Big data analytics, Computational Drug discovery, and Bioinformatics. She is a Life Member of the Indian Society for Technical Education (ISTE).

G. Murugaboopathi received the Undergraduate Degree in Computer Science and Engineering from Madurai Kamaraj University in 2000, the Post Graduate degree in Digital Communication and Network from Madurai Kamaraj University in 2002 and Ph.D in Computer Science and Engineering at Bharath University, Chennai. He has more than 45 publications in National, International Conference and International Journal proceedings. He has more than 15 years of teaching experience. His areas of interest include Wireless Sensor Networks, Bioinformatics. Mobile Communication, Mobile Adhoc Networks, Mobile Computing, Cloud Computing, Network Security, Network and Data Security, Cryptography and Network security. He is currently working as an Associate Professor in the Department of Computer Science and Engineering at Kalasalingam Academy of Research and Education, Tamil Nadu, India.
Journal of Cyber Security and Mobility, Vol. 7_1, 145–160. River Publishers
doi: 10.13052/jcsm2245-1439.7111
This is an Open Access publication. © 2018 the Author(s). All rights reserved.