The Brain Storm Dynamic Technique DBSO-MSA for Efficiently Resolving Multiple Sequence Alignment
Jeevana Jyothi Pujari1 and Kanadam Karteeka Pavan2,*
1Computer Science and Engineering, Acharya Nagarjuna University, Andhra Pradesh, India
2R.V.R & J. C. College of Engineering, Andhra Pradesh, India
E-mail: ajeevanajyothi.pujari@email.com; bkarteeka@yahoo.com
*Corresponding Author
Received 26 October 2021; Accepted 11 January 2022; Publication 17 March 2022
Abstract
Multiple Sequence Alignment (MSA) is a critical step in molecular biology. Different techniques are having been proposed for obtaining optimal alignments, still, there is a need of developing accurate and efficient techniques for optimal sequence alignment. One of the efficient techniques among the swarm optimization families is Brain Storm Optimization Technique based on human social behavior has achieved success in numerous applications. However, population divergence plays a major role in obtaining better solutions for optimization problems. Therefore, high diverged populations obtain optimal results. The multiple sequences alignment is an efficient optimization for dataset analysis but hidden samples do not get tracked by MSA. Therefore DBSO_MSA model requirement is there to crossover limitations of the above model. This paper proposed a dynamic clustered and populated Brain Storm Optimization Algorithm for obtaining more optimal alignment solutions for the Multiple Sequence Alignment problem (DBSO-MSA). The dynamic nature with respect to the number of clusters and population generation at every iteration is incorporated into BSO. The number of solutions and cluster size at each iteration is controlled by the probability variable either it increases or decreases the solution space to explore more diversification in obtaining alignments for the MSA problem. The experiments show DBSO-MSA effectively improves the alignment score on the benchmark sequence datasets compared to the Classical BSO and other evolutionary algorithms.
Keywords: Multiple sequence alignment, brainstorm optimization algorithm, dynamic population, dynamic cluster size.
1 Introduction
Bioinformatics is the most active area of research in the field of computational biology. Sequence alignment is one of the core prerequisites in the area of bioinformatics which guides to identification of one most conserved and similar regions by considering the alignment of two or more DNA/RNA/protein sequences [1]. Alignment of two sequences is termed as pairwise alignment whereas aligning more than sequences is a Multiple Sequence Alignment (MSA) problem. Aligning multiple sequences be a challenging task while considering the variations caused by mutations in the sequences [2]. So MSA belongs to the NP-complete problem [3]. The aligned sequences can applicable in many areas such as constructing phylogenetic evolutionary based trees, predicting protein structure and homology between newly discovered and existing family of sequences [4], foretelling protein mutations [5] and so on. From few decades, various techniques are applied to solve MSA problem, even though it remains as unsolved problem [6].
Local and global MSA are the two forms of MSA. Local alignment prefers block wise search and randomized alignment blocks in the sequences, whereas global alignment attempts to align sequences over their whole length. A wide range of techniques have been developed to perform MSA, an accurate and exact method is dynamic programming (DP) is a mathematical & probabilistic method which simplifies the complex problem by breaking it into simpler and smaller components repeatedly. The most frequently used DP technique for global alignment is Needleman-Wunch algorithm [7], Smith-Waterman technique [8] is for local alignment. These traditional algorithms are best suited to align two sequences, when it came to align more than two sequences with O(Ln) time complexity. To overcome the constraints of the progressing technique, an iterative process has been used to align sequences ideally by maximizing the score, which is a suggested alternate strategy. In an iterative method, solutions are modified across many iterations until they hit a stopping condition or can no longer be improved in terms of their objective function. Many algorithms have been created using an iterative process. One of the iterative approach techniques for aligning numerous sequences ideally is evolution or nature inspired approaches. Computational annealing, Evolutionary algorithms, Dynamical Evolution, and swarm intelligence are the most common inhabitants strategies utilized for MSA.
Literature Survey
In this section a brief discussion of DBSO and MSA related technologies are analyzed through latest published papers. Most of earlier technologies are unable to estimate iterative operation in MSA therefore an advanced technology is required.
| S No | Author | Technique | Key Point |
| 1 | J. Pei, [2008] | Using Iterative refinement and consistency scoring technique | These Servers like Expresso and PROMALS3D have simplified these procedures and made it easier to generate high-quality alignments. |
| 2 | M. Kayed et al. [2020] | Using NestMSA technology | MSA is defined, a new MSA technique named “NestMSA” is proposed, and it is evaluated in two domains: the extraction of web data and the removal of various URLs with identical content (DUST). The PSO optimization method is a model for the proposed approach. I |
| 3 | L. Wang et al. [1994] | Adapting Multiple Sequence Alignment models | To begin, both problems are proven to be MAX SNP-hard and NP-complete, respectively. The degree of difficulty in aligning a tree with a certain phylogeny is also taken into account. |
| 4 | J. C. Gelly et al. [2011] | Utilizing “IPBA” technique | Sequence alignment and superimposed 3D structures using PyMol and Jmol are provided as outputs. There is also an incorporated option for local alignment to find subs-structural similarities. We think the scientific community would benefit much from it since it is a quick and efficient sequence-based’ structural comparison tool. |
In practice, it faces high dimensional problems with the increased number of sequences in MSA also increases computation time and space exponentially. Therefore, heuristic MSA approaches are progressive and iterative techniques used to approximate the optimality of MSA instead of exact approach. Feng and Doolittle [9] developed a widely used progressive alignment algorithm. Progressive technique aligns sequences progressively, that is adding sequences towards the alignment one through one using DP algorithm iteratively. After all, constructing phylogenetic tree to depict the sequence associations. Once a sequence is aligned in early stages that cannot be undone in later phase due to its greedy nature. The misalignments carry forwards to the later stages. The techniques which follows progressive nature are clustal family (X, W, V) [10], MAFFT [11], Kalign [12] and so on.

Figure 1 Multiple sequence alignment.
To defeat the limitations raised in progressive approach, a recommended alternative approach is iterative mechanism has been adopted to align sequences optimally by maximizing the score. In iterative approach the solutions are refined through many iterations either it reaches a stopping criterion or until further improvements cannot be made with respect to their objective function. Many algorithms have been developed using iterative approach. Evolutionary or nature inspired methods are one of the iterative approach algorithms to align multiple sequences optimally. Most population based techniques used for MSA are simulated annealing [13], Genetic algorithm [14], Differential Evolutionary [15], swarm intelligence [16].
One of the socially inspired behavior of humans and recently developed powerful warm intelligence algorithm is Brain Storm Optimization (BSO) algorithm [17]. To deal with the complex issues which are not solved by individual effort, the human brainstorming sessions were conducted, where new ideas are emulated by the current ideas with a horde of people gathered together to find the solution with their conversations and debates shown in Figure 1. Due to its enhanced search space and simplicity suitable for solving many real world engineering applications [18]. In BSO, each solution or individual is regarded as a potential issue solution. A possible solution. Every iteration, all individual ideas are grouped by clustering process and the best solution act as a center of the corresponding group. After all each idea is replaced by the best fitted individual which is generated from the exchange of ideas from one or two selected clusters. Regardless of BSO performance, balancing between exploration and exploitation is a challenging task. Many other versions of the BSO algorithm have been presented to improve its efficacy and performance. To improve their exploratory search and exploitation capacity, the versions ADMBSO [19], An MBSO for addressing restricted optimization problems [20], and [21–23] are employed. However, BSO cannot maintain diverse population to solve many problems. To enhance the performance with the population diversity of BSO and to evade the local optima situation in solving MSA problem efficiently and optimally introduces a new mechanism of dynamic nature with respect to generating individuals and the clustering process. The DBSO algorithm is an intelligent technique to provide information from MSA input. The following input is process to intelligence steps and providing simple solution to complex MSA.
Proposed Dynamic Brain Storm Optimization algorithm (DBSO) presents a strategy of finding optimal sequence alignment by employing dynamic adjustment nature in BSO algorithm. In order to evade local optima situation in producing candidate solutions for MSA problem, dynamically varying the clusters and population size at each iteration is developed in DBSO. In DBSO, an integer encoding solution is used. Due to its integer encoding of candidate solutions as gaps also reduces the computational complexity and the length of the ideas. For evaluating the efficacy of proposed algorithm, experiments were conducted on benchmark dataset and compared with the results obtained from GA and BSO [24] techniques on MSA.
The following are the categories for the research paper: Section 2 may be used to describe the MSA issue. The third section introduces the BSO method. The suggested Dynamic BSO for solving MSA, as well as solution encoding and associated fitness scoring function, are described in depth in Section 4. The results of the proposed DBSO algorithm are presented in Section 5 compared with the existing techniques. Finally, Section 6 provides the findings and potential directions for improvement.
2 MSA Scheme
Multiple Sequence Alignment (MSA) is a technique to arrange multiple sequences to anticipate and highlight similarities, evaluate the function and organisation of sequence families and is also an essential stage for many developed biological forecasts. The alignment of a pair of sequences is a pair of wise alignment, whereas numerous sequences align more than two sequences. The length and quantity of sequences depend on the alignment of various sequences. Given a set of n sequences S {s,s,…,s} over an alphabet where length of each sequence defined on {l,l,…,l} respectively. A new alphabet is introduced where ‘‘ is a gap character made of either insertion or deletion in biological sequences. composed of 4 characters in DNA, or in case of proteins it’s a collection of 20-characters. For the given sequences finding out the aligned sequences S {s,s,…,s} by fulfilling the constraints of , all sequences possesses the same length, removing gaps in Inserting gaps in S should be equal to S, and no column in the alignment entirely made up of gaps.
3 Original BSO
BSO gains more popularity in solving engineering and scientific applications due to the philosophy of human brain storming. In BSO model is a population-based technique, where an idea from individual human brain taken as a solution to critical problem. All the ideas generated from the group of people are gathered and can follows the procedure at each iteration. The procedure involved in BSO follows.
3.1 Idea Generation
Initially N randomly generated solutions or individual ideas to solve the problem in a search space. The fitness of N solutions is calculated through their objective function.
3.2 Clustering
Next step is to disrupt N solutions as randomly selected C clusters based on their individual fitness and C is lesser than N. The clustering operation refine the search space and the best fitted solution can be recorded as a center of corresponding cluster group. Replacing of cluster center can be done through the chosen random replaceable probability of pr_replace. The replacement of the cluster centre with a random notion is regulated by pr replace.
3.3 Selection of Individuals for Mutation
To maintain the diversity in population, BSO generates new ideas either selection of one idea or two ideas from one or two clusters respectively. The selection of individuals from the cluster or clusters depends on the probability variable prone. Choosing of centers from clusters or individuals from clusters are according to the selection probabilities of prone center or orthocenter. The ideas are selected as specified in Equation (1).
| (1) | |
| (2) | |
| (3) |
3.4 New Individual Generation
After selecting ideas from the selected clusters or clusters, additional ideas are produced using Equation (2). X and Xl are the newly developed and chosen ideas from the clusters correspondingly. N(0,1) is the Gaussian circulation value with the mean 0 and the variance is 1. and § à is step length, adding weights to the Gaussian spreading function computed using Equation (3). Maxitr is the maximum amount of iterations provided by the user, and curtitr is the present quantity of iterations in the algorithm. logsig() is the sigmod function used. The slope adjustment can be done through k. The random value lies in between 0 and 1 and selected randomly. When the fitness of novel knowledge is greater than the current idea then the current idea is replaced by new idea. This updating procedure is done for all ideas in the iteration. At each iteration for all the individual ideas the procedure is repeated from (c) to (d). The entire process is repeated until maximum iterations reached or the optimal solution can be found.
3.5 DBSO MSA
In this section, we discuss about DBSO which solves MSA problem efficiently by exploring more search space solutions by its population diversity technique. Inspired by the adaptive and dynamic population size mechanisms proposed in different nature inspired algorithm [22, 25, 26] the proposed Dynamic Brain Storm Optimization algorithm (DBSO) has been designed to solve MSA. In DBSO the population diversity can be enhanced by automatically incrementing and decrementing the population size based on a probability variable that effects the number of clusters for obtaining optimal alignment solutions. The population growth cost can be ranged using a randomly distributed value between which can scale down and enlarging the size of the population. The dynamic cluster size also varies with the newly generated population size with the new factor rate r as [0, 0.2].
3.6 Individual Representation
Each Brain Storm Optimization algorithm population is named an idea and provides a potential alignment explanation for the specific MSA problem. Initially consider, n sequences as input and determine their lengths. Choose the maximum length sequence as a reference sequence for global alignment. For a given set of n classifications , and the length of the sequences are li {l,l,…l} i {1,2,…n} for n sequences. The matrix can be defined as collection of sequences; whose rows represent categorizations of specified length is represented in A.
Where ALG defined by taking gap character in addition to the dna/rna/protein sequences from the alphabet . i.e. U{}. Determining the gaps in each sequence needs to calculate largest sequence length, and adding more number of gaps up to 15% of the original largest sequence length. All the sequences in ALG have the same length jrand(max(li), max(li)*15%) and varied. The matrix ALG should not contain a single row or column of having all characters as gaps. Another constraint added in order to obtain the solution is maximizing the match score and the number of gaps should be minimized in the alignment solution set. Initially generate N ideas as encoding solutions consists of gap incidence locations in the sequences. number of gaps in each sequence depends on their length and varies and can be shown in Equation (4). After the gaps are incorporated into sequences the total aligned length is same for the sequences.
| (4) |
3.7 Scoring Objective Function
In iterative based algorithms the selection of objective functions are the primary measures for validating solutions. Various scoring schemes are available to evaluate the alignments in MSA problem. The preferred scoring objective functions are Similarity score (SS) [27], Sum of Pair score (SOP), Column Score (COS), Weighted Sum-of-Pairs Score (WSP) [28]. The aligned sequences in the proposed DBSO are evaluated by considering SOP scoring function in account. The purpose is to exploit the SOP fitness determination of produced solutions at each iteration. SOP score can be formed as the test alignment composed of n number of sequences and l columns as the length of the sequences, the ith column in the aligned solution is termed as {S, S,… S}. For each pair S, S residues with the specified condition P. The score for the ith column is expressed as Si in Equation (5). Where n considers quantity of classifications in the alignment process, l and lr are the lengths of columns in the arrangement and orientation alignment respectively.
| (5) |
4 Proposed Methodology
The proposed DBSO algorithm performance can be enhanced by dynamically changing the population size in the search space, giving chance to explore and to evade the situation of arising local optima. The dynamic population size is a feasible solution to improve the quality of solutions with respect to their fitness value. The steps followed are: initially generate N solutions encoded as gap occurrence positions in each sequence as a matrix. The potential aligned solutions are assessed as maximizing functions via the sum-of-pairs (sop) objective function. The population can be sorted according to the highest fitness value and disrupt all the individuals as nc number of clusters. Make the best one as cluster center. The selection of cluster centers or individual populations for generating new population entirely depends on the selection probabilities. Replace the old person with the new person depending on fitness. For all the population the above said procedure is repeated in that iteration. The task of choosing appropriate dynamic population size in-between the depends over a pre specified random decision variable . Introducing randomness into the proposed algorithm gives more optimal score than the original BSO. The size of dynamic population can be estimated through the following equation specified in (6). The dynamic population size can be adding current population size to the increase or diminish the relative growth rate parameter according to the selected value of within the
| (6) |
If (N N) that is the new population size is more than the current one, then elitism is used to preserve the current ideas and the remaining ideas (N N) are generated randomly to make the required number of population in the coming generation. In the other case where the new population size is lower than the current population size, the best fitted individuals will be promoted into the next generation and the worst fitted individuals will be culled from the next generation. the elitism and culling will not be included where the condition (N N) occurs. Cluster size will also be affected with the new population size and re clustering has been taken place to exchange more ideas which also explores more divergence for exploitation purpose. The proposed DBSO algorithm is discussed and as shown.
The Algorithm 4 is explaining about DBSO modeling for MSA, in this at input stage parameters are to be initializing. The population and event evolutions are used to get completed information about MSA sing loop and whiles concept data had been individually dividing.
5 Experiment Results
This section describes the experiments conducted on the sequence alignment benchmark dataset OXBench [29] to evaluate the optimal alignment performance and investigation of the parameter modifications in the proposed algorithm DBSO. The optimality can be shown by aligning randomly chosen sequences from the benchmark dataset using proposed algorithm over other evolutionary algorithms such as Genetic Algorithm (GA) and Brain Storm Optimization (BSO) algorithm. Initially the values pertaining to the specified parameters yields optimal alignment score are shown in Table 1. The GA and BSO algorithms are most prominent to estimating gap length and Max scores of MSA. The positions and sequences are generating scores according to scores getting MSA allignment.
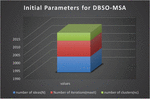
Table 1 Initial parameters for DBSO-MSA algorithm
| S.no. | Parameters | Values |
| 1 | Number of ideas (N) | 2000 |
| 2 | Number of iterations (maxit) | 10 |
| 3 | Number of clusters (nc) | 5 |
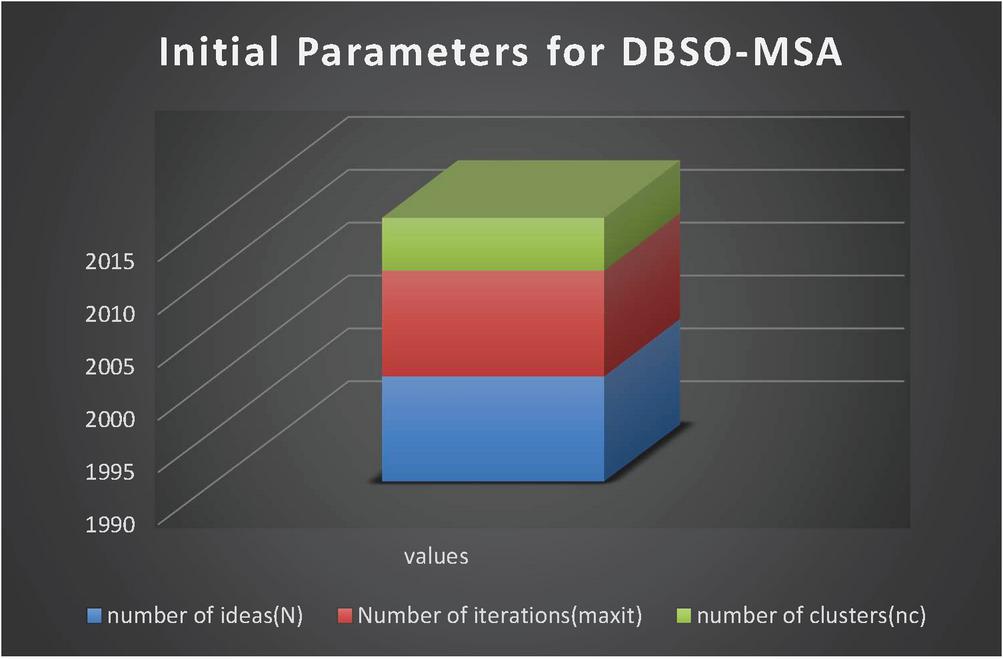
Figure 2 Initial parameters for DBSO-MSA.
The above Figure 2 and Table 1 is clearly explains about DBSO-MSA initial parameters in this many functionalities like number of ideas, number of iterations and clusters to be taken are explained. The following elements are getting analyzed by proposed algorithm and find out the easy step for analysis.
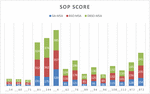
Table 2 SoP score for DBSO with MSA
| Sl.N | Sequences | Sequence No | GA-MSA | BSO-MSA | DBSO-MSA |
| 1 | 3 | __54 | 33 | 52 | 52 |
| 2 | 3 | __60 | 34 | 44 | 52 |
| 3 | 3 | __71 | 23 | 47 | 47 |
| 4 | 6 | __85 | 191 | 312 | 324 |
| 5 | 5 | __144 | 175 | 362 | 374 |
| 6 | 7 | __4 | 320 | 357 | 385 |
| 7 | 6 | __62 | 85 | 116 | 124 |
| 8 | 4 | __76 | 121 | 178 | 186 |
| 9 | 3 | __89 | 42 | 85 | 102 |
| 10 | 4 | __94 | 95 | 125 | 136 |
| 11 | 3 | __96 | 56 | 89 | 75 |
| 12 | 6 | __108 | 115 | 132 | 132 |
| 13 | 3 | __112 | 85 | 101 | 110 |
| 14 | 3 | _4t2 | 128 | 156 | 165 |
| 15 | 7 | _8t2 | 154 | 235 | 241 |
From the observe results in Table 2, it illustrated that the proposed DBSO algorithm is significantly performs well over GA and BSO. On the other case, BSO and DBSO performs equally well in yielding optimal score for three sequence sets such as __54, __71 and __108. In one case only BSO obtains more score than DBSO. The best optimal alignment results for algorithms can be shown in bold. It shows the efficiency of the proposed algorithm with respect to the Equation (4) with increasing and decreasing population size as well as choosing cluster size dynamically during evolution.
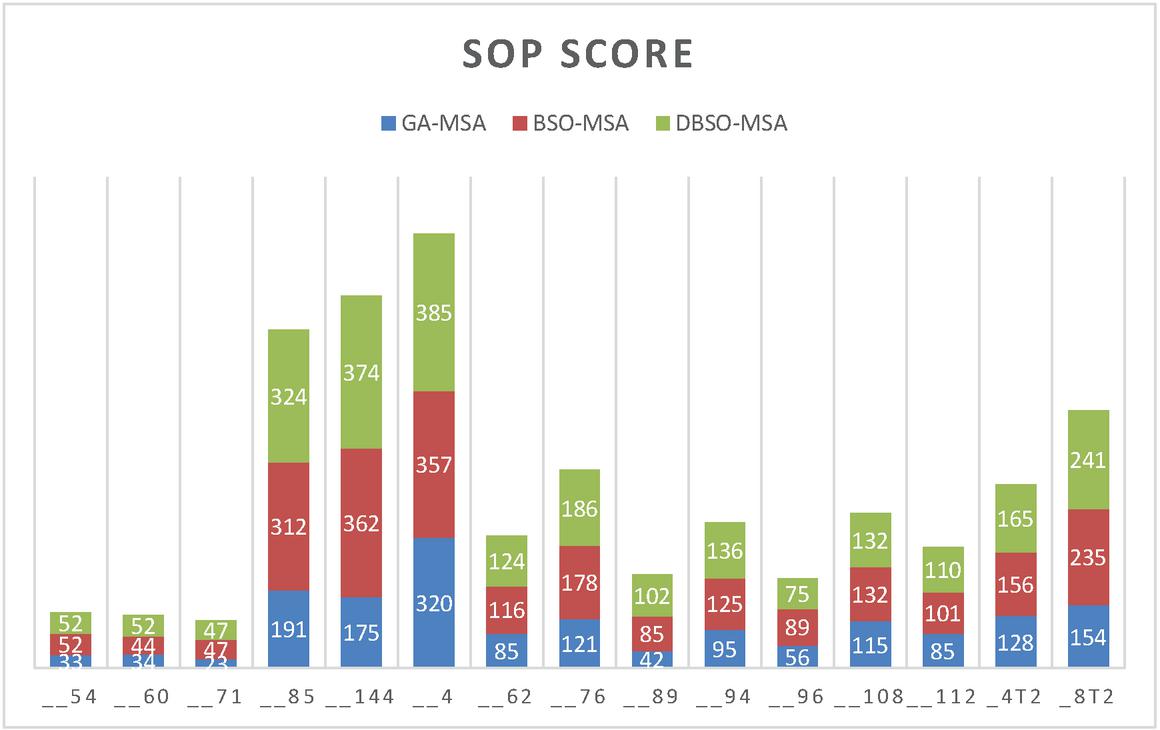
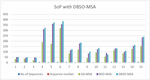
Figure 3 Performance comparison of SoP score DBSO-MSA earlier models.
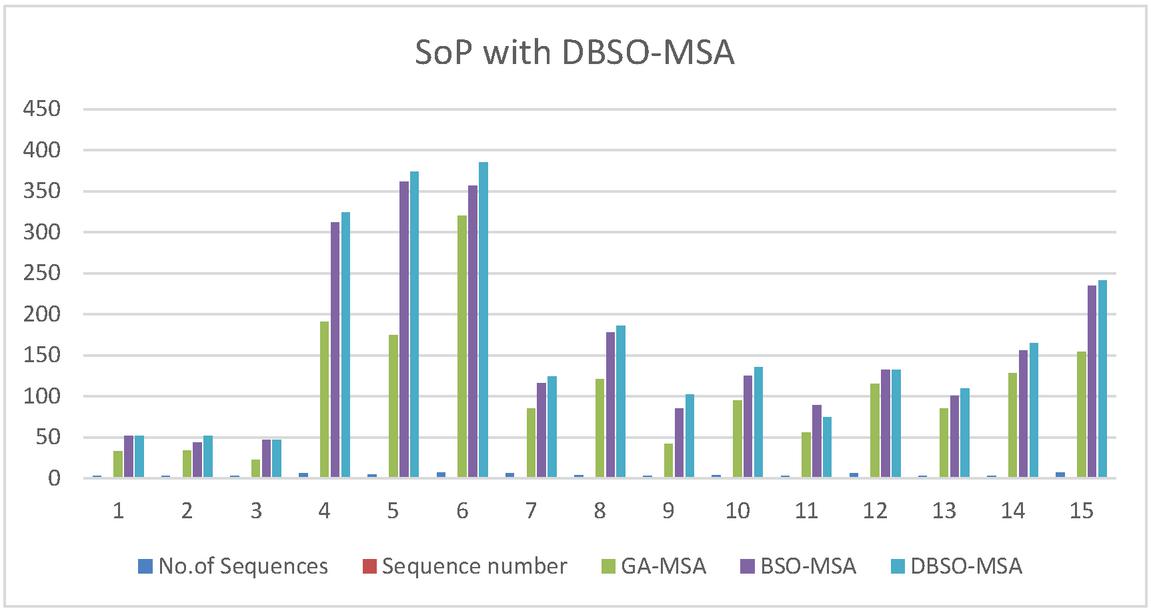
Figure 4 Comparative results of sop score for DBSO-MSA along with GA and BSO Algorithms.
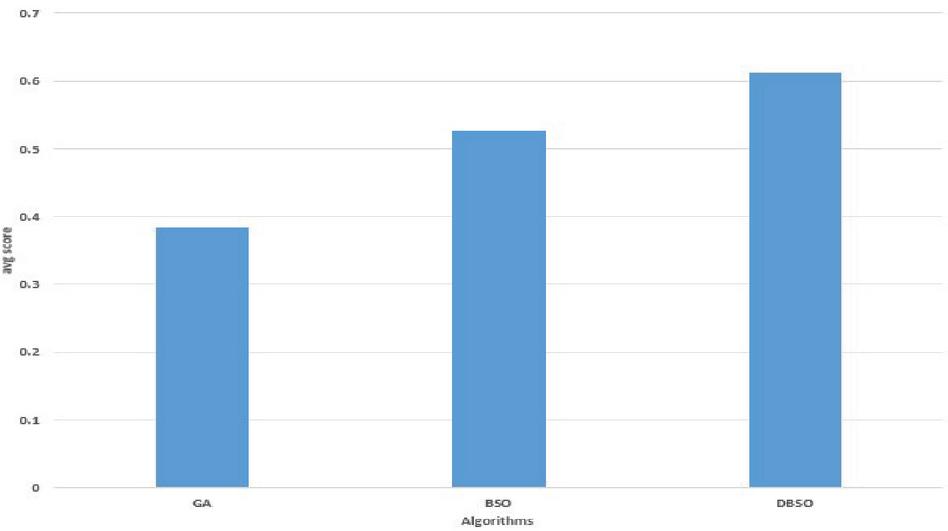
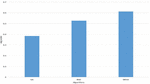
Figure 5 Performance of proposed DBSO with GA and BSO algorithms.
The comparison of average sop scores of DBSO and various algorithms are illustrated in Figures 1 and 2. It shows that the average sop scores for DBSO algorithm for alignment score has better performance than the comparative algorithms of GA and BSO. The DBSO-MSA algorithm has the ability to produce more optimal alignment score compared to GA and BSO due to its dynamic parameter adjustment in each iteration and evades the local optima situation. Figure 3 show the overall performance efficiency of DBSO, GA and BSO algorithms. It also indicates that the DBSO outperforms both GA and BSO algorithms for obtaining alignment score. The DBSO achieves 0.8% improvement over BSO whereas over GA, DBSO achieves 22.2% more with respect to sop score alignment.
The above Figure 3 is clearly explains about average sop score alignment results for DBSO-MSA along with GA and BSO algorithms. In this proposed model, DBSO providing accurate analysis and giving optimal alignment score. According to DBSO scores, classification of MSA had been done, moreover targets are equivalent the residues and proteins. The following reason can improves the 22.2% accuracy score.
Figure 4 is clearly explaining about sop score for DBSO-MSA along with GA and BSO Algorithms. In this proposed model attains more improvement compared earlier models.
The above Figure 5 is clearly explains about Average optimal score with various algorithms like GA, BSO and DBSO algorithms. In this DBSO algorithm is more improved at avg optimal score compared to GA and BSO.
Conclusions
This proposed work suggested Dynamic Brain Storm Optimization (DBSO) algorithm inspired by the social behavior of human ideas. The ideas are used as solutions to our Multiple sequence alignment problem and also guarantee in obtaining maximum optimal score in alignment. Dynamic nature in cultivated into the population and optimal number of clusters also improves the solution space by exploring more ideas and clustering improves the exploitation by exchanging ideas at each iteration. Both are reducing the chances for getting into local optima then evades premature convergence. Moreover, the most optimal score is obtained through proposed DBSO algorithm for MSA compared to the other GA and BSO algorithms. We concluded as, due to the simplistic and dynamic nature of DBSO algorithm gives efficient results for solving MSA problem. In future we will concentrate on solving MSA optimally by examining multiple objectives for the lengthy sequences. The Scoring P[S2 S1] p [S4, S5] p [S6 S4] 1 t t 2t can providing optimal score with better performance with DBSO.
References
[1] J. Pei, “Multiple protein sequence alignment,” Curr. Opin. Struct. Biol., vol. 18, no. 3, pp. 382–386, 2008, doi: 10.1016/j.sbi.2008.03.007.
[2] M. Kayed and A. A. Elngar, “NestMSA: a new multiple sequence alignment algorithm,” J. Supercomput., vol. 76, no. 11, pp. 9168–9188, 2020, doi: 10.1007/s11227-020-03206-0.
[3] L. Wang and T. Jiang, “On the Complexity of Multiple Sequence Alignment,” J. Comput. Biol., vol. 1, no. 4, pp. 337–348, 1994, doi: 10.1089/cmb.1994.1.337.
[4] J. C. Gelly, A. P. Joseph, N. Srinivasan, and A. G. De Brevern, “IPBA: A tool for protein structure comparison using sequence alignment strategies,” Nucleic Acids Res., vol. 39, no. SUPPL. 2, pp. 18–23, 2011, doi: 10.1093/nar/gkr333.
[5] S. Hicks, D. A. Wheeler, S. E. Plon, and M. Kimmel, “Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed,” Hum. Mutat., vol. 32, no. 6, pp. 661–668, 2011, doi: 10.1002/humu.21490.
[6] D. A. Morrison, “Multiple Sequence Alignment is not a Solved Problem,” 2018, [Online]. Available: http://arxiv.org/abs/1808.07717.
[7] S. B. Needleman and C. D. Wunsch, “A general method applicable to the search for similarities in the amino acid sequence of two proteins,” J. Mol. Biol., vol. 48, no. 3, pp. 443–453, Mar. 1970, doi: 10.1016/0022-2836(70)90057-4.
[8] T. F. Smith and M. S. Waterman, “Identification of common molecular subsequences,” J. Mol. Biol., vol. 147, no. 1, pp. 195–197, Mar. 1981, doi: 10.1016/0022-2836(81)90087-5.
[9] D.-F. Feng and R. F. Doolittle, “Progressive sequence alignment as a prerequisitetto correct phylogenetic trees,” J. Mol. Evol., vol. 25, no. 4, pp. 351–360, Aug. 1987, doi: 10.1007/BF02603120.
[10] R. Chenna et al., “Multiple sequence alignment with the Clustal series of programs,” Nucleic Acids Res., vol. 31, no. 13, pp. 3497–3500, 2003, doi: 10.1093/nar/gkg500.
[11] K. Katoh and D. M. Standley, “MAFFT multiple sequence alignment software version 7: Improvements in performance and usability,” Mol. Biol. Evol., vol. 30, no. 4, pp. 772–780, 2013, doi: 10.1093/molbev/mst010.
[12] T. Lassmann and E. L. L. Sonnhammer, “Kalign - An accurate and fast multiple sequence alignment algorithm,” BMC Bioinformatics, vol. 6, pp. 1–9, 2005, doi: 10.1186/1471-2105-6-298.
[13] J. Kim, S. Pramanik, and M. J. Chung, “Multiple sequence alignment using simulated annealing,” Bioinformatics, vol. 10, no. 4, pp. 419–426, 1994, doi: 10.1093/bioinformatics/10.4.419.
[14] C. Gondro and B. P. Kinghorn, “A simple genetic algorithm for multiple sequence alignment.,” Genet. Mol. Res., vol. 6, no. 4, pp. 964–82, Oct. 2007, [Online]. Available: https://pubmed.ncbi.nlm.nih.gov/18058716/.
[15] R. C. Addawe, J. M. Addawe, M. R. K. Sueno, and J. C. Magadia, “Differential evolution-simulated annealing for multiple sequence alignment,” J. Phys. Conf. Ser., vol. 893, no. 1, 2017, doi: 10.1088/1742-6596/893/1/012061.
[16] S. Lalwani, R. Kumar, and N. Gupta, “Efficient two-level swarm intelligence approach for multiple sequence alignment,” Comput. Informatics, vol. 35, no. 4, pp. 963–985, 2016.
[17] Y. Shi, “Brain storm optimization algorithm,” Lect. Notes Comput. Sci. (including Subser. Lect. Notes Artif. Intell. Lect. Notes Bioinformatics), vol. 6728 LNCS, no. PART 1, pp. 303–309, 2011, doi: 10.1007/978-3-642-21515-5\_36.
[18] S. Cheng, Q. Qin, J. Chen, and Y. Shi, “Brain storm optimization algorithm: a review,” Artif. Intell. Rev., vol. 46, no. 4, pp. 445–458, 2016, doi: 10.1007/s10462-016-9471-0.
[19] Y. Yang, Y. Shi, and S. Xia, “Advanced discussion mechanism-based brain storm optimization algorithm,” Soft Comput., vol. 19, no. 10, pp. 2997–3007, 2015, doi: 10.1007/s00500-014-1463-x.
[20] A. Cervantes-Castillo and E. Mezura-Montes, “A modified brain storm optimization algorithm with a special operator to solve constrained optimization problems,” Appl. Intell., vol. 50, no. 12, pp. 4145–4161, 2020, doi: 10.1007/s10489-020-01763-8.
[21] Y. Yu, S. Gao, Y. Wang, Z. Lei, J. Cheng, and Y. Todo, “A Multiple Diversity-Driven Brain Storm Optimization Algorithm with Adaptive Parameters,” IEEE Access, vol. 7, pp. 126871–126888, 2019, doi: 10.1109/ACCESS.2019.2939353.
[22] J. Liu, H. Peng, Z. Wu, J. Chen, and C. Deng, “Multi-strategy brain storm optimization algorithm with dynamic parameters adjustment,” Appl. Intell., vol. 50, no. 4, pp. 1289–1315, 2020, doi: 10.1007/s10489-019-01600-7.
[23] S. Cheng, Y. Shi, Q. Qin, Q. Zhang, and R. Bai, “Population Diversity Maintenance In Brain Storm Optimization Algorithm,” J. Artif. Intell. Soft Comput. Res., vol. 4, no. 2, pp. 83–97, 2014, doi: 10.1515/jaiscr-2015-0001.
[24] P. Jeevana Jyothi, K. P. K, S. M. Raiyyan, and T. Rajasekhar, “MSA: An Application of Brain Storm Optimization Algorithm,” SSRN Electron. J., 2020, doi: 10.2139/ssrn.3646174.
[25] J. Teo, “Exploring dynamic self-adaptive populations in differential evolution,” Soft Comput., vol. 10, no. 8, pp. 673–686, 2006, doi: 10.1007/s00500-005-0537-1.
[26] H. Wang, S. Rahnamayan, and Z. Wu, “Adaptive Differential Evolution with variable population size for solving high-dimensional problems,” 2011 IEEE Congr. Evol. Comput. CEC 2011, pp. 2626–2632, 2011, doi: 10.1109/CEC.2011.5949946.
[27] K. Chellapilla and G. B. Fogel, “Multiple sequence alignment using evolutionary programming,” Proc. 1999 Congr. Evol. Comput. CEC 1999, vol. 1, no. February 1999, pp. 445–452, 1999, doi: 10.1109/CEC.1999.781958.
[28] J. D. Thompson, F. Plewniak, and O. Poch, “A comprehensive comparison of multiple sequence alignment programs,” Nucleic Acids Res., vol. 27, no. 13, pp. 2682–2690, 1999, doi: 10.1093/nar/27.13.2682.
[29] G. P. S. Raghava, S. M. J. Searle, P. C. Audley, J. D. Barber, and G. J. Barton, “OXBench: A benchmark for evaluation of protein multiple sequence alignment accuracy,” BMC Bioinformatics, vol. 4, pp. 1–23, 2003, doi: 10.1186/1471-2105-4-47.
Biographies

Jeevana Jyothi Pujari, is working as Associate Professor in Computer Science and Engineering at Vasireddy Venkatadri Institute of Technology. Published seven research papers in International Journals and five papers in International Conferences. Pursuing PhD in Acharya Nagarjuna University in the area of research is Bio-informatics.

Kanadam Karteeka Pavan, is working as Professor in RVR & JC College of Engineering. She completed her PhD from Acharya Nagarjuna University. Her areas of research are Data Mining and Bio-informatics. She is life time member for CSI. She published 25 papers in International Journals and 15 papers in International Conferences. She completed one research project, published one book and organized two workshops.
Journal of Mobile Multimedia, Vol. 18_4, 1191–1210.
doi: 10.13052/jmm1550-4646.18411
© 2022 River Publishers