Protein Prediction using Dictionary Based Regression Learning
T. Sudha Rani1,*, A. Yesu Babu2 and D. Haritha3
1Department of Computer Science and Engineering, Aditya Engineering College, ADB Road, Aditya Nagar, Surampalem, Andhra Pradesh, India
2Department of Computer Science and Engineering, Sir C R Reddy College of Engineering, Eluru, Andhra Pradesh, India
3Department of Computer Science and Engineering, Jawaharlal Nehru Technological University Kakinada, India
E-mail: proftsudharani1430@gmail.com
*Corresponding Author
Received 29 August 2022; Accepted 21 November 2022; Publication 29 April 2023
Abstract
Research Objectives: Molecular genetic data is managed by the information technology known as bioinformatics. Major concept involved in bioinformatics is a protein sequence. Amino acids bonded with peptide bond constitute the sequence of Protein and it is very essential to lead life. To predict sequence of amino acid, primary sequence obtains amino sequence folding and structures prediction.
Research Novelty: In this manuscript, dictionary based regression learning and fuzzy genetic algorithm is proposed for protein prediction from structural analysis (DRL-FGA-PD-SA). In this input data are taken from Kaggle domain dataset. The extraction of protein features from given data is made through Kernel Matrix (KM) which extracts composition of amino acids, composition of dipeptide, composition of pseudo-amino-acid, composition of functional domain and distance-based features. Then fuzzy based genetic algorithm (FGA) update the selected features for classification of protein and the features are clustered. Finally, dictionary based regression learning (DRL) predicts the class of protein with conversion of values either 0’s or 1’s.
Research Conclusions: The proposed method is executed on MATLAB. Here evaluation metrics as sensitivity, precision, f-measure, specificity, accuracy and error rate are outlined. Then the performance of the proposed DRL-FGA-PD-SA method provides 22.08%, 24.03%, 34.76% higher accuracy, 23.34%, 26.45%, 34.44% higher precision, compared with the existing systems such assdeep learning methods in protein structure prediction (FFNN-RNN-PD-SA), deep learning technique for protein structure prediction and protein design (DNN-PD-SA) and improved protein structure prediction using potentials from deep learning (DNN-SGDA-PD-SA) respectively.
Keywords: Protein sequence, amino acid sequence, kernel matrix, fuzzy based genetic algorithm, dictionary based regression learning.
Abbreviation
In the computational biology field, the most difficult task is the protein prediction from its sequence of amino acids. Protein tertiary is predicted by the most known approaches [1, 2]. Template-based modelling (TBM) with protein threading and homology modelling. Query protein are predicted by TBM with the solved structures of one or multiple template’s alignment. The protein structures are predicted by TBM with better percentage along with the PDB growth [3] For example, templates responsible in PDB are present in CASP14 58 out of 107 test domains and in CASP13 67 out of 112 test domains. Templates which are highly similar are not present in protein that is to be predicted, three major difficulties faced by TBM are: best template selection, an accurate sequence-template alignment are constructed, and from the alignment, 3D models are constructed [4–6].
Primary, secondary, tertiary, and quaternary are the four classes categorize proteins with respect to the structure of polypeptide [7]. For the non-homologous sequences of protein, next-generation sequencing (NGS) produces low accuracy, and time-consuming results and because of the technique NGS, it is very difficult to analyze the behavior of protein [8–12]. Thus, to handle the large set, deep learning algorithms are applied for designing the computational protein with the probability of 20 amino acids present in protein prediction [13–16]. Genetic information are supported highly by 3D protein structure which is effectively defined by predicting the protein, as the 3D structure of protein is available limitedly and due to this experimental biologist suffers a lot. Design of drug, protein function discovery, and mutations interpreted in structural genomics are the biological process applied in the numerous applications obtained from the 3D structure of protein predicted from the sequence of amino acids [17–19].
In this manuscript, dictionary based regression learning and fuzzy genetic algorithm for protein prediction from structural analysis is proposed. In this the input data are taken from Kaggle domain dataset. The extraction of protein features from given data is made through KM which extracts composition of amino acids, composition of dipeptide, composition of pseudo-amino-acid, composition of functional domain and distance-based features. Then FGA update the selected features for classification of protein and the features are clustered. Finally, DRL predicts the class of protein with conversion of values either 0’s or 1’s.
The main contribution of this manuscript is précised as follow,
• In this manuscript, dictionary based regression learning and fuzzy genetic algorithm is proposed for protein prediction from structural analysis (DRL-FGA-PD-SA).
• In this the input data are taken from Kaggle domain dataset [20].
• The extraction of protein features from given data is made through kernel Matrix (KM) [21] which extracts composition of amino acids, composition of dipeptide, composition of pseudo-amino-acid, composition of functional domain and distance-based features.
• Then fuzzy based genetic algorithm (FGA) [22] updates the selected features for classification of protein and the features are clustered.
• Finally, dictionary based regression learning (DRL) [23] predicts the class of protein with conversion of values either 0’s or 1’s.
• The proposed system is executed on MATLAB. Here evaluation metrics as sensitivity, precision, f-measure, specificity, accuracy and error rate are outlined.
• Then the performance of the proposedCov-19-MPNN-GRF-CTI system is compared with existing approaches such as FFNN-RNN-PD-SA, DNN-PD-SA and DNN-SGDA-PD-SA respectively.
Remaining manuscript is defined as, Section 2 reviews literature survey, Section 3 clarifies proposed Methodology, Section 4 shows results and discussion and Section 5 completes the manuscript.
2 Related Works
Among numerous research works related to protein prediction, a few recent research works are reviewed below.
Torrisi et al., [24] have introduced deep learning approaches for predicting protein structure. One-dimensional and two-dimensional Protein Structure Annotations are discussed in this basic feed-forward Neural Networks and Recurrent Neural Networks prediction. Statistical approaches ranged from simple in the early days to the computationally costly, extremely advanced Deep Learning algorithms of last decade. The evolution of the databases on which these algorithms are built, and how this has impacted the ability to use knowledge about evolution and co-evolution to make better predictions. It provides higher accuracy with lower recall.
Pearce and Zhang [25] have introduced deep neural networks for protein constraint prediction, end-to-end model training has considerably improved the accuracy of protein structure prediction, mostly explaining the issue at fold level used for single-domain proteins. The field of protein design has likewise seen remarkable growth. The prominent examples have illustrated the stored information of neural-network models. It can be used to create sophisticated functional proteins. Thus, incorporating deep learning systems into various stages of protein folding and design methods has a transformational impact on both domains. It provides higher accuracy with maximum error rate.
Senior et al., [26] have introduced a neural network to predict the distances among the sets of protein’s residues which expressed additional about structure than contact predictions. By this information constructed a potential of mean force that were exactly explain about the protein’s shape. Then find that a simple gradient descent algorithm was optimized the resulting potential, for realize the structures without the necessity for complex sampling procedures. It delivers higher recall with lower accuracy.
Kryshtafovych et al., [27] have presented the critical assessment approaches of protein structure prediction (CASP) for estimating three-dimensional (3D) protein structure through amino acid sequence. The arduous blind testing of approaches and the evaluation of the findings by independent assessors are key components. This model had already demonstrated its ability to present resolutions for difficult crystal structures, and it had far-reaching implications for the remainder of structural biology. It provides higher estimation model of accuracy with maximum error rate.
Yang et al., [28] have reported an improved protein structure prediction method that makes use of projected interresidue orientations. A deep residual- Convolutional network takes multiple sequence alignments (MSA) as input and outputs information on linked distances and orientations of the entire protein residue pairs. According to the network outputs, a quick Rosetta model building technique based on restricted minimization with distance and orientation constraints was developed. It has a lesser accuracy with a reduced error rate.
3 Proposed Methodology
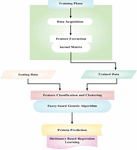
In this manuscript, dictionary based regression learning and fuzzy genetic algorithm is proposed for protein prediction from structural analysis. The block diagram of proposed DRL-FGA-PD-SA method is given in Figure 1. The libraries and protein data bank (PDB) datasets [15] are introduced. The extraction of protein features from given data is made through kernel Matrix (KM) which extracts composition of amino acids, composition of dipeptide, composition of pseudo-amino-acid, composition of functional domain and distance-based features. Then fuzzy based genetic algorithm (FGA) updates the selected features for classification of protein and the features are clustered. Finally, dictionary based regression learning (DRL) predicts the class of protein with conversion of values either 0’s or 1’s. The detailed discussion regarding dictionary based regression learning and fuzzy genetic algorithm for protein prediction from structural analysis are given below.
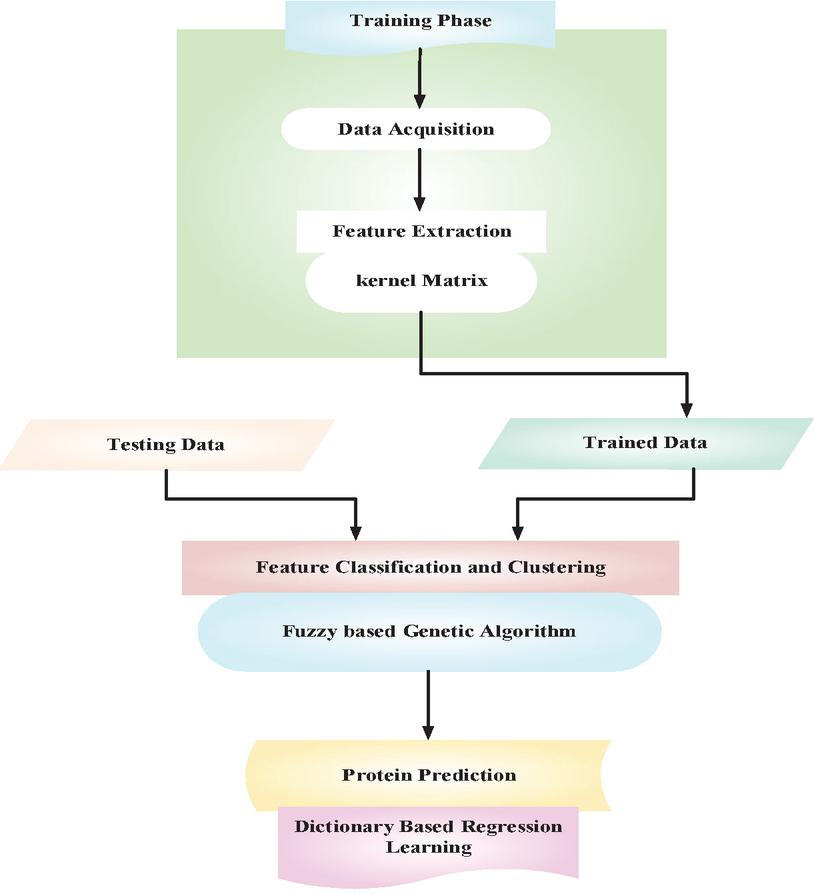
Figure 1 Block diagram of proposed DRL-FGA-PD-SA method.
3.1 Data Acquisition
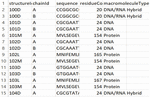
The Kaggle domain for structural protein sequences dataset is used in this investigation. The dataset may be found at: https://www.kaggle.com/shahir/protein-data-set\#pdbdataseq.csv. The PDB Protein Research Collaborator for Structural Bioinformatics (RCSB) [15] used to generate this protein dataset. The collection is separated into two sub-sets: the first provides data on protein Meta that has details on protein identification are given into Figure 2 and the second part has protein structure sequences which are shown in Figure 3. The protein’s “structure ID” attribute used to organise the two datasets. The first dataset contains 1,41,000 rows and 14 columns, whereas the second contains 4,67,000 rows and 5 columns. This raw dataset is then filtered by deleting any empty or superfluous fields. Lastly, for more efficient processing, we only considered the first 10,000 rows.
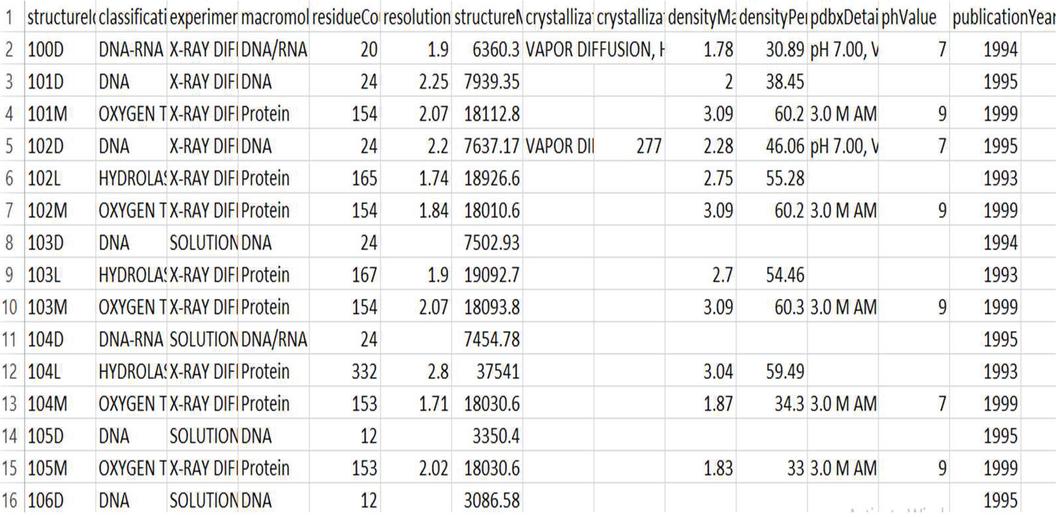
Figure 2 Screenshot of first part of structural protein sequences dataset.
Figure 3 Screenshot of second part of structural protein sequences dataset.
3.2 Kernel Matrix Based Feature Extraction
The input data are given to kernels Matrix for extraction of features. The relevant and representative samples in the kernels Matrix selection procedure calculate the significance scores with a single pass over the input data set. The calculated significance scores are given with the valued information to choose different examples based on distance metric specified by the kernel function. Individual chains present in the receptor and ligand , obtains the feature representation which are complex in the dataset. The Protein structure’s sequence-based attributes are modelled by using different kernel representations and explicit features. The evolutionary relationship among protein is modelled by using position specific scoring matrix features (PSSM). For obtaining this representation, the PSSM of a provided sequence of protein was used. For every chain of protein, complex PSSM was obtained by utilizing kernels Matrix for three iterations contrary to the database of non-redundant protein having threshold value. The sequence of protein is represented as s by the PSSM with average columns, in the feature representation. In this 20-dimensional feature vector is obtained using Equation (1) as follows,
| (1) |
From Equation (1), indicates the PSSM column equivalent to the residue in . The sequences with various lengths, which sharing the common portion are compared by using Local alignment kernel. In this method, each and every input sequence’s local alignment contributions are added by kernel. Statistically, the score of local alignment among the protein sequence is expressed in Equation (2),
| (2) |
here indicates the local alignment’s score, kernel sensitivity is controlled by the parameter . The value of is equal to 0.1 when for higher values of kernel score. The complex-level kernel was developed to predict the complex sequence-based kernels. This complex-level kernel is obtained from the two complexes with the average of individual chains values of kernel function. It is expressed using Equation (3),
| (3) |
where and indicates the complex kernel, and indicates the chain level kernel for two chains from the two complexes. At last, kernel Matrix function extracts composition of amino acids, composition of dipeptide, composition of pseudo-amino-acid, composition of functional domain and distance-based features.
3.3 Fuzzy Based Genetic Algorithm for Feature Classification and Feature Clustering
After completing the feature extraction process, the extracted protein features are classified and clustered using fuzzy based genetic algorithm (FGA). Here, the extracted protein features are given to fuzzy based genetic algorithm (FGA) and it is used to classify those features. In a extensive sense, Multi-objective optimization algorithms are a unique tools for exploring the space of design to Pareto-optimal solutions on a reasonable time. Moreover the proposed fuzzy based genetic algorithm (FGA) enables procedure of decision making through expansively searching the potential regions for a superior classification. The proposed FGA integrates the fuzzy decision making with the average linkage-based hierarchical clustering algorithm to present the manageable clustering. In fuzzy technique the minimizing criterion is defined in Equation (4) as follows,
| (4) |
where every membership in fuzzy with the weighing component is represented by , indicates the cluster centre and denotes the membership value. FGA is proceeded by the process of iteration calculating the values of membership , and as cluster centres, is presented in the Equation (5),
| (5) | ||
| (6) |
where classification is carried out by and clustering is carried out by which is known as indices of cluster validity. Xie–Beni (XB) index is the index given as the fraction of the total variation to the minimal split-up of the clusters. And it is computed using Equation (7) as follows,
| (7) |
The minimum separation of the clusters is computed using Equation (8),
| (8) |
Then expression of XB index is given by Equation (9),
| (9) |
where lower values of and higher values of produced XB index with lower values when there is good and compact partitioning. To attain the proper clustering values, XB index is needed to be minimized. XB index does not work with any simple methods, not like which uses simple strategies in FGA. So that is used in clustering. This XB index is not reduced monotonically with the cluster numbers, not like , due to this, it is not utilized for varying number of clusters. XB index is efficient for the suitable number of clusters. Suitable cluster centre set is evolved by using fuzzy based genetic algorithm (FGA). In encoding strategy, are used to encode cluster centre by a chromosome, the number of dimensions is represented by . From the dataset cluster centres are selected randomly for a chromosome initialization. Then, is the size of population where it is repeated times. Extraction of the encoded cluster centres are performed first. Cluster close to the centre are assigned with every data point. Each and every tie is arbitrarily determined. Each new cluster centre is calculated by mean value of the cluster assigned. The original centre encoded by chromosome is replaced by this new centre. The strategy of proportional selection is implemented by fuzzy based genetic algorithm (FGA). Using the probability of fixed crossover, it will have single-point crossover and this is responsible for the mutation of chromosome. The gene position value in mutation is disconcerted by a little quantity. Continuing the process, with the highest iteration number, then the output obtained for the problem of clustering is the best stringand then classification of protein is performed using the updated selected features.
3.4 Dictionary Based Regression Learning for Protein Prediction
Then clustered features are provided to dictionary based regression learning (DRL) for the prediction of protein class. It purposes to cater the unified and the exhaustive protein predictions to all clustered and classified data with protein annotations. Additionally, it provides the biological community immediate access to outcomes from predictors chosen not just for their availability and convenience of use. Sparsely represent able sequences of target protein assumed to be in the patches. Few patches in the dictionary are linearly combined with every patch coding in the DRL-based patch method. Thebest global over-complete dictionary was found in this method, representing few patches in the dictionary vectors (atoms) are linearly combined with every patch coding (atoms). The linear combination coefficients are predicted by the process of sparse coding. The problem based on DRL-based patch method is solved by the Equation (10) as follows,
| (10) |
where original data are represented by , clustered data are denoted by , and denotes the data index, indicates the operator, in which the patch is extracted, denotes the patch-based dictionary, expresses the linear combination used to approximate every patch, the vector entries which are non-zero are counted by and sparsity level preset parameter is given by limiting the nonzero entry number as maximum in and it is calculated using Equation (11),
| (11) |
Dictionary and coefficients in the clustered data patch set is trained by the solution of the above equation. The dictionary based regression learning (DRL)is used for replacing by the data with observed knowledge. For the calculations done practically, scaling ambiguity is avoided by constraining dictionary columns as unit norm. The output data is obtained from the solution of 1st order derivatives with the available dictionary and , which is expressed mathematically in Equation (12) as follows,
| (12) |
The pre-calculated dictionary or global dictionary derived is used to define the entire process ofdictionary based regression learning (DRL) processing is given in Equation (13),
| (13) |
From Equation (13), is represented as tolerance parameter. The intensive loss of is used to perform the regression and complexity of this model is controlled. The prediction of protein binding affinity is estimated using Equation (14),
| (14) |
where indicates the input protein sequence and represents the secondary sequence neighbourhood. And it is computed using Equation (15) as follows,
| (15) |
Such that for all,
| (16) |
where the margin is controlled by , the margin violation extent is captured by and for a training sample provided and penalty is denoted by violations, indicates the linear function and denotes the radial basis regression function. At last, dictionary based regression learning (DRL) predicts the class of protein with conversion of values either 0’s or 1’s.
4 Results and Discussion
This section describes dictionary based regression learning and fuzzy genetic algorithm for protein prediction from structural analysis. The entire implementation of proposed scheme is done in MATLAB. The PC configuration for the implementation is set as Intel Core i7-4790 3.60 GHz CPU with 32 GB RAM. Here, the performance metrics are analysed to validate the performance of proposed approach. Then, the proposed approach is analysed with other different existing methods, such as FFNN-RNN-PD-SA [19], DNN-PD-SA [20] and DNN-SGDA-PD-SA [21] respectively.
4.1 Performance Metrics
To validate the performance of proposed model, the performance metrics, viz accuracy, precision, recall and error rate are evaluated.
4.1.1 Accuracy
It is a ratio of exact predictions to a total count of proceedings in dataset. And it is determined as the following Equation (17),
| (17) |
where indicates the true positive rate, represents true negative rate, denotes false positive rate and indicates false negative rate.
4.1.2 Precision
It is defined as the classifier’s capacity to calculate the protein class in the absence of any conditions. The equation provides it (18),
| (18) |
4.1.3 Recall
It is defined as the ratio of the number of properly classified records to the total number of modified events. The equation provides it (19),
| (19) |
4.2 Simulation Result
Tables 1–4 shows the simulation results of the dictionary based regression learning and fuzzy genetic algorithm for protein prediction from structural analysis. The performance metrics are analysed to validate the performance of proposed system. The performance of proposed DRL-FGA-PD-SA system is compared with existing systems, as FFNN-RNN-PD-SA, DNN-PD-SA and DNN-SGDA-PD-SA respectively.
Table 1 depicts the Accuracy analysis. Here the proposed DRL-FGA-PD-SA method attains 18.87%, 16.64% and 15.45% higher accuracy for hydrolase protein; 13.47%, 11.84% and 25.59%, higher accuracy for transferase protein; 16.38%, 11,53% and 25.56% higher accuracy for oxidoreductase protein; 16.29%, 13.75% and 26.85% higher accuracy for immune system protein; 14.45%, 15.45% and 25.74%, better accuracy for transcription protein; 18.36%, 24.65% and 17.57% higher accuracy for signalling protein; 10.84%, 15.54% and 18.25%, higher accuracy for lyase protein; 12.87%, 10.98% and 23.98% higher accuracy for transport protein; 13.25%, 23.36% and 18.56% higher accuracy for protein binding; 19.83%, 13.35% and 27.54% higher accuracy for structural genomic, unknown function protein are compared with existing FFNN-RNN-PD-SA, DNN-PD-SA and DNN-SGDA-PD-SA methods respectively.
Table 1 Accuracy comparison
| Accuracy (%) | ||||
| FFNN-RNN- | DNN- | DNN-SGDA- | DRL-FGA- | |
| Class | PD-SA | PD-SA | PD-SA | PD-SA (Proposed) |
| Hydrolase | 83 | 82 | 80 | 94 |
| Transferace | 78 | 76 | 74 | 95 |
| Oxidoreductace | 73 | 75 | 76 | 96 |
| Immune system | 80 | 84 | 86 | 97 |
| Transcription | 79 | 78 | 77 | 98 |
| Signaling protein | 72 | 73 | 74 | 99 |
| Lyase | 76 | 77 | 78 | 97 |
| Transport protein | 79 | 78 | 81 | 98 |
| Protein Binding | 88 | 78 | 77 | 97 |
| Structural genomic unknown function | 83 | 87 | 88 | 99 |
Table 2 Precision comparison
| Precision (%) | ||||
| FFNN-RNN- | DNN- | DNN-SGDA- | DRL-FGA- | |
| Class | PD-SA | PD-SA | PD-SA | PD-SA (Proposed) |
| Hydrolase | 71 | 81 | 72 | 94 |
| Transferace | 73 | 82 | 74 | 95 |
| Oxidoreductace | 77 | 87 | 77 | 96 |
| Immune system | 76 | 79 | 78 | 97 |
| Transcription | 81 | 76 | 81 | 96 |
| Signaling protein | 84 | 78 | 88 | 98 |
| Lyase | 80 | 75 | 87 | 99 |
| Transport protein | 79 | 74 | 84 | 98 |
| Protein Binding | 78 | 79 | 76 | 97 |
| Structural genomic unknown function | 86 | 83 | 83 | 99 |
Table 2 depicts the Precision analysis. Here the proposed DRL-FGA-PD-SA method attains 14.74%, 20.63% and 17.98% higher precision for hydrolase protein; 14.98%, 24.45% and 18.34% higher precision for transferase protein; 16.86%, 13.87% and 20.87% higher precision for oxidoreductase protein; 18.98%, 26.23% and 15.85% higher precision for immune system protein; 11.89%, 25.67% and 16.87% better precision for Transcription protein; 19.98%, 14.56% and 29.75% higher precision for signaling protein; 17.97%, 27.98% and 13.76% higher precision for lyase protein; 23.23%, 15.98% and 19.55% higher precision for transport protein; 15.67%, 16.76% and 23,34% higher precision for protein binding; 14.98%, 11.87% and 27.76% higher precision for structural genomic, unknown function protein are compared with existing FFNN-RNN-PD-SA, DNN-PD-SA and DNN-SGDA-PD-SA methods respectively.
Table 3 Recall comparison
| Recall (%) | ||||
| FFNN-RNN- | DNN- | DNN-SGDA- | DRL-FGA- | |
| Class | PD-SA | PD-SA | PD-SA | PD-SA (Proposed) |
| Hydrolase | 71 | 75 | 70 | 92 |
| Transferace | 73 | 85 | 76 | 94 |
| Oxidoreductace | 78 | 75 | 81 | 96 |
| Immune system | 75 | 84 | 74 | 98 |
| Transcription | 81 | 73 | 79 | 91 |
| Signaling protein | 85 | 72 | 73 | 93 |
| Lyase | 88 | 84 | 77 | 95 |
| Transport protein | 72 | 82 | 87 | 97 |
| Protein Binding | 77 | 84 | 88 | 99 |
| Structural genomic unknown function | 76 | 86 | 84 | 98 |
Table 4 Error rate comparison
| Error Rate (%) | ||||
| FFNN-RNN- | DNN- | DNN-SGDA- | DRL-FGA- | |
| Class | PD-SA | PD-SA | PD-SA | PD-SA (Proposed) |
| Hydrolase | 17 | 18 | 20 | 6 |
| Transferace | 12 | 24 | 26 | 5 |
| Oxidoreductace | 27 | 25 | 24 | 4 |
| Immune system | 20 | 16 | 14 | 3 |
| Transcription | 21 | 22 | 23 | 2 |
| Signaling protein | 28 | 27 | 26 | 1 |
| Lyase | 24 | 28 | 22 | 3 |
| Transport protein | 21 | 22 | 19 | 2 |
| Protein Binding | 12 | 22 | 23 | 3 |
| Structural genomic unknown function | 17 | 13 | 12 | 1 |
Table 3 depicts the Recall analysis. Here the proposed DRL-FGA-PD-SA method attains 14.65%, 15.76% and 23.65% higher recall for hydrolase protein; 17.87%, 26.65% and 13.78% higher recall for transferase protein; 10.89%, 16.98% and 17.87% higher recall for oxidoreductase protein; 11.73%, 17.34% and 27.35% higher recall for immune system protein; 18.56%, 25.87% and 14.78% better recall for Transcription protein; 17.75%, 23.76% and 12.90% higher recall for signalling protein; 16.89%, 27.89% and 13.78% higher recall for lyase protein; 17.89%, 29.89% and 15.67% higher recall for transport protein; 18.90%, 14.65% and 15.87% higher recall for protein binding; 19.78%, 24.93% and 11.89% higher recall for structural genomic, unknown function protein are compared with existing FFNN-RNN-PD-SA, DNN-PD-SA and DNN-SGDA-PD-SA methods respectively.
Table 4 depicts the Error rate analysis. Here the proposed DRL-FGA-PD-SA method attains 14.98%, 17.83% and 27.46% lower error rate for hydrolase protein; 15.76%, 16.89% and 19.78% lower error rate for transferase protein; 12.78%, 23.87% and 17.87% lower error rate for oxidoreductase protein; 13.76%, 18.98% and 27.98% lower error rate for immune system protein; 17.98%, 15.87% and 25.87% lower error rate for Transcription protein; 18.78%, 25.78% and 11.78 lower error rate for signalling protein; 16.89%, 27.89% and 13.78% lower error rate for lyase protein; 10.87%, 14.87% and 18.83% lower error rate for transport protein; 13.35%, 17.56% and 15.73% lower error rate for protein binding; 19.78%, 16.98% and 12.67% lower error rate for structural genomic, unknown function protein are compared with existing FFNN-RNN-PD-SA, DNN-PD-SA and DNN-SGDA-PD-SA methods respectively.
5 Conclusions
In this, dictionary based regression learning and fuzzy genetic algorithm is successfully implemented for protein prediction. Then the proposed DRL-FGA-PD-SA method is implemented in MATLAB and the effectiveness is estimated with the aid of several evaluation metrics such as accuracy, Error rate, recall and precision. Here the proposed DRL-FGA-PD-SA method attains 41.26%, 73.10% and 24.12% higher recall 31.14%, 14.88% and 33.27% lower error rate compared with existing methods like FFNN-RNN-PD-SA, DNN-PD-SA and DNN-SGDA-PD-SA respectively. In the future, accuracy may be enhanced by taking into account more than four vectors, as well as other factors like pH, Molecular weight, and other components, which may yield more information on family group and also contain more characters to permit for greater interaction among amino acids.
References
[1] T. Siebenmorgen, M. Zacharias, ‘Computational prediction of protein–protein binding affinities. Wiley Interdisciplinary Reviews’, Computational Molecular Science, vol. 10, no. 3, p. e1448, 2020.
[2] M. Al Quraishi, ‘Machine learning in protein structure prediction’, Current opinion in chemical biology, vol. 65, pp. 1–8, 2021.
[3] M. Torrisi, G. Pollastri, Q. Le, ‘Deep learning methods in protein structure prediction’, Computational and Structural’ Biotechnology Journal, vol. 18, pp. 1301–1310, 2020.
[4] J. Pereira, A.J. Simpkin, M.D. Hartmann, D.J. Rigden, R.M. Keegan, A.N. Lupas, ‘High-accuracy protein structure prediction in CASP14’, Proteins: Structure, Function and Bioinformatics, vol. 89, no. 12, pp. 1687–1699, 2021.
[5] M. Zeng, F. Zhang, F.X. Wu, Y. Li, J. Wang, M. Li, ‘Protein–protein interaction site prediction through combining local and global features with deep neural networks’, Bioinformatics, vol. 36, no. 4, pp. 1114–1120, 2020.
[6] C. Chen, Q. Zhang, B. Yu, Z. Yu, P.J. Lawrence, Q. Ma, Y. Zhang, ‘Improving protein-protein interactions prediction accuracy using XG Boost feature selection and stacked ensemble classifier’, Computers in Biology and Medicine, vol. 123, p. 103899, 2020.
[7] X. Yang, S. Yang, Q. Li, S. Wuchty, Z. Zhang, ‘Prediction of human-virus protein-protein interactions through a sequence embedding-based machine learning method’, Computational and structural biotechnology journal, vol. 18, pp. 153–161, 2020.
[8] Y. Duan, R. Coreas, Y. Liu, D. Bitounis, Z. Zhang, D. Parviz, M. Strano, P. Demokritou, W. Zhong, ‘Prediction of protein corona on nanomaterials by machine learning using novel descriptors’, NanoImpact, vol. 17, p. 100207, 2020.
[9] P. Rajesh, FH. Shajin, ‘Optimal allocation of EV charging spots and capacitors in distribution network improving voltage and power loss by Quantum-Behaved and Gaussian Mutational Dragonfly Algorithm (QGDA)’, Electric Power Systems Research, vol. 194, pp. 107049, 2021.
[10] P. Rajesh, FH. Shajin, BN. Kommula, ‘An efficient integration and control approach to increase the conversion efficiency of high-current low-voltage DC/DC converter’, Energy Systems, pp. 1–20, 2021.
[11] FH. Shajin, P. Rajesh, MR. Raja, ‘An Efficient VLSI Architecture for Fast Motion Estimation Exploiting Zero Motion Prejudgment Technique and a New Quadrant-Based Search Algorithm in HEVC’, Circuits, Systems and Signal Processing, vol. 41, no. 3, pp. 1751–74, 2022.
[12] FH. Shajin, P. Rajesh, S. Thilaha, ‘Bald eagle search optimization algorithm for cluster head selection with prolong lifetime in wireless sensor network, ‘Journal of Soft Computing and Engineering Applications, vol. 1, no. 1, pp. 7, 2020.
[13] S.C. Pakhrin, B. Shrestha, B. Adhikari, D.B. Kc, ‘Deep learning-based advances in protein structure prediction’, International Journal of Molecular Sciences, vol. 22, no. 11, p. 5553, 2021.
[14] B. Niu, C. Liang, Y. Lu, M. Zhao, Q. Chen, Y. Zhang, L. Zheng, K.C. Chou, ‘Glioma stages prediction based on machine learning algorithm combined with protein-protein interaction networks’, Genomics, vol. 112, no. 1, pp. 837–847, 2020.
[15] R. Chowdhury, N. Bouatta, S. Biswas, C. Floristean, A. Kharkar, K. Roy, C. Rochereau, G. Ahdritz, J. Zhang, GM. Church, PK. Sorger, ‘Single-sequence protein structure prediction using a language model and deep learning’, Nature Biotechnology, pp. 1–7, 2022.
[16] M. Zeng, F. Zhang, F.X. Wu, Y. Li, J. Wang, M. Li, ‘Protein–protein interaction site prediction through combining local and global features with deep neural networks’, Bioinformatics, vol. 36, no. 4, pp. 1114–1120, 2020.
[17] A.H. Mahmoud, M.R. Masters, Y. Yang, M.A. Lill, ‘Elucidating the multiple roles of hydration for accurate protein-ligand binding prediction via deep learning’, Communications Chemistry, vol. 3, no. 1, pp. 1–13, 2020.
[18] K. Sato, M. Akiyama, Y. Sakakibara, ‘RNA secondary structure prediction using deep learning with thermodynamic integration’, Nature communications, vol. 12, no. 1, pp. 1–9, 2021.
[19] A.W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A.W. Nelson, A. Bridgland, H. Penedones, ‘Improved protein structure prediction using potentials from deep learning’, Nature, vol. 577, no. 7792, pp. 706–710, 2020.
[20] https://www.kaggle.com/shahir/protein-data-set\#pdb\_data\_seq.csv
[21] P. Hajibabaee, F. Pourkamali-Anaraki, M.A. Hariri-Ardebili, ‘Kernel matrix approximation on class-imbalanced data with an application to scientific simulation’, IEEE Access, vol. 9, pp. 83579–83591, 2021.
[22] A. Rain, M.E. Saritac, ‘HydroPower Plant Planning for Resilience Improvement of Power Systems using Fuzzy-Neural based Genetic Algorithm’, arXiv preprint arXiv: 2106.12042, 2021.
[23] P. Goyal, P. Benner, ‘Discovery of nonlinear dynamical systems using a Runge–Kutta inspired dictionary-based sparse regression approach’, Proceedings of the Royal Society A, vol. 478, no. 2262, pp. 20210883, 2022.
[24] M. Torrisi, G. Pollastri, Q. Le, ‘Deep learning methods in protein structure prediction’, Computational and Structural Biotechnology Journal, vol. 18, pp. 1301–1310, 2020.
[25] R. Pearce, Y. Zhang, ‘Deep learning techniques have significantly impacted protein structure prediction and protein design’, Current opinion in structural biology, vol. 68, pp. 194–207, 2021.
[26] A.W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A.W. Nelson, A. Bridgland, H. Penedones, ‘Improved protein structure prediction using potentials from deep learning’, Nature, vol. 577, no. 7792, pp. 706–710, 2020.
[27] A. Kryshtafovych, T. Schwede, M. Topf, K. Fidelis, J. Moult, ‘Critical assessment of methods of protein structure prediction (CASP)—Round XIV’, Proteins: Structure, Function and Bioinformatics, vol. 89, no. 12, pp. 1607–1617, 2021.
[28] J. Yang, I. Anishchenko, H. Park, Z. Peng, S. Ovchinnikov, D. Baker, ‘Improved protein structure prediction using predicted interresidue orientations’, Proceedings of the National Academy of Sciences, vol. 117, no. 3, pp. 1496–1503, 2020.
Biographies

T. Sudha Rani, received B.Tech degree in IT from R.VR & JC college of Engineering affiliated to Nagarjuna University, Guntur, and Andhra Pradesh in 2005. M.Tech Degree in CSE from JNTUA, Anantapur, Andhra Pradesh in 2010. She is currently working as Associate Professor, Department of Computer Science and Engineering, Aditya Engineering College, ADB Road, Aditya Nagar, Surampalem, Andhra Pradesh, India and Pursuing Ph.D at JNTUK, Kakinada. Her area of Research are Bioinformatics and Data Mining.

A. Yesu Babu, Currently working as a Professor, Department of Computer Science and Engineering, Sir C R Reddy College of Engineering, Eluru, Andhra Pradesh, India. He is having 31 years of Academic, Research & Academic Administration experience. Published 43 Research Papers in International journals and 6 chapters. Reviewer of Research publications for premier publishing groups like Springer, Elsevier, Inderscience and a number of SCOPUS and SCI indexed journals.

D. Haritha, She is working as Associate Professor in Computer science and Engineering Department at Jawaharlal Nehru Technological University Kakinada, India. She has 17 years of experience. She guided 50 M.Tech students and 15 MCA students for their project. Her research interest is on Image Processing, Data Structures, Software Engineering and Networking. She published 12 research papers in international journals. She published 11 research papers in international conferences.
Journal of Mobile Multimedia, Vol. 19_4, 963–984.
doi: 10.13052/jmm1550-4646.1942
© 2023 River Publishers